
Abstract
Introduction:
Hydronephrosis is characterized by substantial loss of tubules and affects renin secretion in the kidney. However, whether alterations of angiotensin-converting enzyme (ACE), ACE2 and Mas receptor in the heart are observed in hydronephrosis is unknown. Thus, we assessed these components in hydronephrotic mice treated with AT1 receptor blockade and ACE inhibitor.
Materials and methods:
Hydronephrosis was induced by left ureteral ligation in Balb/C mice except sham-operated animals. The levels of cardiac ACE, ACE2 and Mas receptor were measured after treatment of losartan or enalapril.
Results:
Hydronephrosis led to an increase of ACE level and a decrease of ACE2 and Mas receptor in the heart. Losartan decreased cardiac ACE level, but ACE2 and Mas receptor levels significantly increased in hydronephrotic mice (p < 0.01). Enalapril increased ACE2 levels (p < 0.01), but did not affect Mas receptor in the heart. Plasma renin activity (PRA) and Ang II decreased in hydronephrotic mice, but significantly increased after treatment with losartan or enalapril.
Conclusions:
Hydronephrosis increased cardiac ACE and suppressed ACE2 and Mas receptor levels. AT1 blockade caused sustained activation of cardiac ACE2 and Mas receptor, but ACE inhibitor had the limitation of such activation of Mas receptor in hydronephrotic animals.
Introduction
Chronic kidney disease such as hydronephrosis is a severe worldwide problem and causes an increase in cardiovascular risk.1–5 The renal vasoactive system controlling blood pressure and the renin-angiotensin system (RAS) was postulated to be involved in the hypertension implicated in hydronephrosis.6–8 Hydronephrotic hypertension has often been explained to be caused by the RAS activated in the kidney leading to high blood pressure.9–11 In a previous study, we demonstrated that hydronephrosis due to unilateral ureter obstruction causes an increase in the synthesis of renin in the kidney.12–14 Recently, evidence has shown that acute kidney injury causes cardiac remodeling and increases the expression of angiotensin-converting enzyme 2 (ACE2).15–17 This implicates reciprocal interactions between the heart and kidney for cardiovascular regulation, mediated by the RAS. Cardiovascular diseases are commonly associated with an imbalance within the RAS. However, to date, whether cardiovascular regulation through the local RAS is altered in hydronephrosis has yet to be investigated. Therefore, the present study was to assess the regulation of levels of ACE, ACE2 and Mas receptor in the heart in Balb/C mice, with a hydronephrotic kidney induced by left ureteral ligation. This study was designed to evaluate these parameters after treatment with ACE inhibitor and angiotensin (Ang) type I (AT1) blockade. Since the inhibition of vasoconstrictor effects of Ang II with ACE inhibitors or AT1 blockade relates to beneficial renal and cardiac effects in patients with kidney disease,18,19 we have also investigated the effects of ACE inhibition and AT1 blockade on blood pressure, plasma renin activity (PRA), Ang I and Ang II in hydronephrosis.
Materials and methods
Animals and experimental protocol
Sixty male Balb/C mice, aged 10 weeks, were used in this study. The experiments were conducted according to the National Research Council’s guidelines and were approved by the Taishan Medical University Ethics Committee. All mice weighing 25–30 g were subjected to left ureteral ligation except the sham-operated group (n = 15). The surgical procedure of the left ureteral ligation was performed as described previously.12,13 Sham-operated mice underwent a similar operation without the ureteral ligation. Mice were maintained on a standard laboratory diet and had free access to tap water. Four weeks after the operation the left kidney was hydronephrotic.12–14 Following the ureteral ligated surgery, animals were randomly allocated to a vehicle (n = 15) or losartan at a dose of 30 mg kg−1 (Los, n = 15) or enalapril at 20 mg kg−1 (Ena, n = 15) by daily oral gavage for four weeks.
Blood pressure was measured by the tail-cuff method using photoelectric volume oscillometry (ALC-NIBP Analysis System, Aoer Biotech, Shanghai), as described previously.13,14
At four weeks of treatment, animals were sacrificed by intraperitoneal pentobarbital sodium overdose (100 mg kg−1). At postmortem, a blood sample was taken from the abdominal aorta. Plasma was separated from each blood sample by microcentrifuge. Heart tissue was removed and weighed. A whole heart was cut longitudinally and snap-frozen in isopentane/dry ice for immunohistochemical studies. A piece of each tissue was preserved at −80ºC for reverse transcription-polymerase chain reaction (RT-PCR) and Western blot analysis.
PRA, Ang I and Ang II assays
PRA, Ang I and Ang II were measured by a radioimmunoassay using a commercial kit (Puer Weiye Biotech, Beijing) based on methods described previously.12–14
Measurement of ACE, ACE2 and Mas receptor messenger RNA (mRNA)
Total RNA was extracted from the heart using TRIzol (Invitrogen, NY, USA) according to the manufacturer’s protocol. RT-PCR was carried out using a PCR Reagent Kit (TaKaRa, Tokyo, Japan) as described previously. 14 Housekeeping gene β-actin was used as an internal standard. The results were normalized to β-actin mRNA quantitated from the same samples. The primers used in this study are as follows: ACE forward 5’-TAACTCGAGTGCCGAGGTG-3’ and reverse 5’-CCAGCAGGTGGCAGTCTT-3’; ACE2 forward 5’-CTTCAGCACTC TCAGCAG ACA-3’ and reverse 5’-CAACTTCCTCCTCACATAGGC-3’; Mas forward 5’-ACTGTCGGGCGGTCATCATC-3’ and reverse 5’-GGTGGAGAA AAGCAAGGAGA-3’; β-actin forward 5’-GTCGTACCACTGGCATTGTG-3’ and reverse 5’-TCTCAGCTGTGGTGGTGAAG-3’. Densitometric analysis of PCR was performed using the Digital Imaging System (Bio-Rad, Hercules, CA, USA).
Western blot analysis
Protein from heart tissue was extracted and subjected to Western blot analysis as reported previously. 20 Thirty µg of protein was loaded per lane, separated in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were probed using primary antibodies against ACE, ACE2 and Mas receptor (all from Santa Cruz Biotech Inc, Dallas, TX, USA), respectively, followed by horseradish peroxidase-conjugated secondary antibody for two hours at room temperature. Chemoluminescence was detected with an enhanced chemilumescent (ECL) kit (Pierce, Boston, MA, USA) and subsequent exposure to Hyperfilm (Amersham, Piscataway, NJ, USA). The amount of ACE, ACE2 and Mas receptor was expressed relative to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
ACE2 immunohistochemistry
Sections of unfixed hearts were embedded in PolyFreeze Tissue Freezing Medium (Polysciences, Warrington, PA, USA) and snap-frozen in isopentane/dry ice as described previously. 11 Briefly, cryostat sections, 7 µm thick, were cut and fixed in 4% paraformaldehyde for 30 minutes at 4°C. The sections were incubated with the dilution of primary antibody against ACE2 (Santa Cruz Biotech Inc, USA) at dilution of 1:200 in phosphate-buffered saline (PBS) solution. After washing with PBS, they were incubated with peroxidase-conjugated anti-rabbit immunoglobulin (Ig)G (goat polyclonal, 1:5000) for one hour at 4°C. The color was developed with 0.05% 3, 3’-diaminobenzidine tetrahydrochloride (DAB) and 0.01% H2O2. Quantitative analyses of ACE2 expression within the heart were performed using a quantitative image analysis system (Metamorph, CA, USA).
Statistical analysis
All data are expressed as the mean ± SEM. Statistical significance of ACE, ACE2, Mas receptor, PRA, Ang I, Ang II and MAP data were determined with one-way analysis of variance (ANOVA) in SPSS followed by least significant difference (LSD) post hoc tests. Differences were considered statistically significant at a value of p < 0.05.
Results
General data
After left ureteral ligation, mean arterial pressure (MAP) did not differ among groups. At the end of four weeks of experimental procedures, AT1 receptor blockade losartan decreased MAP from 106 ± 5 mmHg to 93 ± 4 mmHg (p < 0.05, Table 1), and ACE inhibitor enalapril decreased MAP from 108 ± 5 mmHg to 95 ± 3 mmHg (p < 0.05) in ureteral ligated mice, respectively.
Body, heart weight, MAP, plasma angiotensins in four groups of mice.

MAP: mean arterial pressure; Ang: angiotensin; BW: body weight; HW: heart weight; Sham: sham-operated; Hy: hydronephrosis; Los: losartan; Ena: enalapril. ap < 0.05, bp < 0.01. n: number of mice in each group.
At postmortem, any liquid in the left hydronephrotic kidney was removed and both the right and left kidneys were weighed. In the sham-operated mice, the right and left kidney weight was 0.20 ± 0.03 g and 0.21 ± 0.04 g, respectively. However, in mice after ureteral ligation the left kidney weight was 0.11 ± 0.01 g and was much lighter than the contralateral kidney (0.28 ± 0.04 g, p < 0.01), indicating that the left kidney was hydronephrotic and the right kidney was enlarged compensatorily. However, there was no difference in body and heart weight between groups (Table 1). Light microscopic observation confirmed that the tubules in the hydronephrotic kidney (Figure 1(b)) disappeared, differing from those in the control kidney (Figure 1(a)).
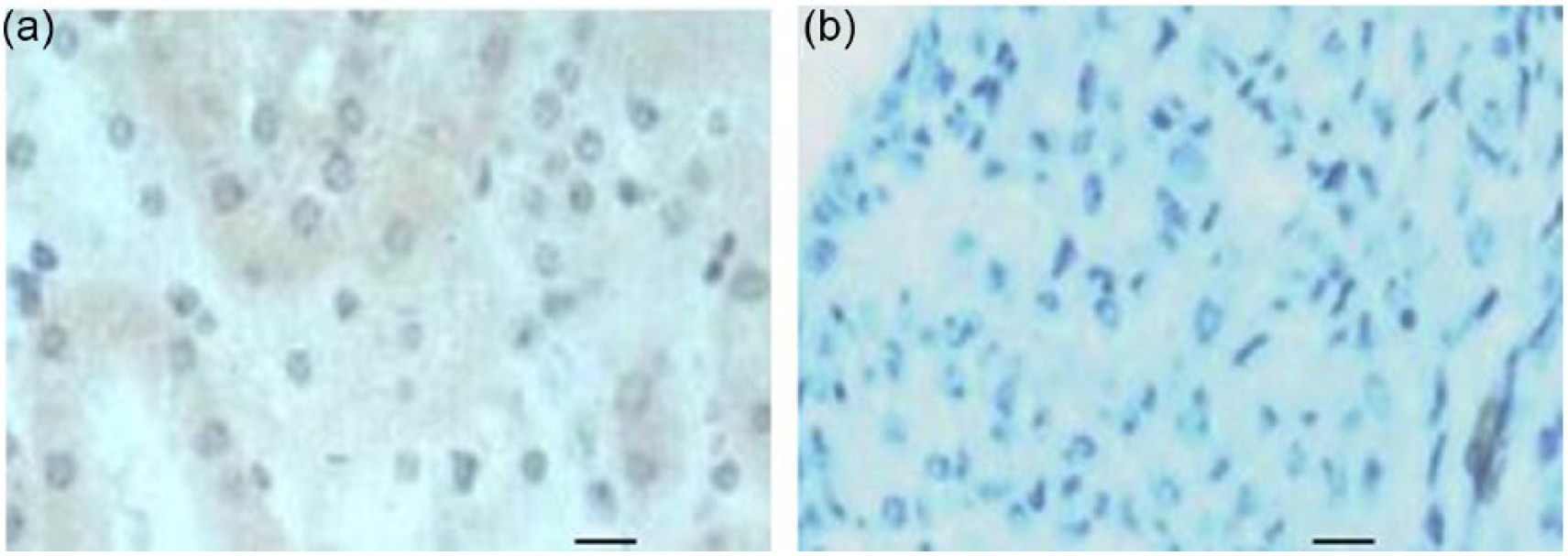
Serial 3 µm paraffin sections of normal (a) and hydronephrotic (b) kidneys in mice. In the normal kidney the tubules were intact while they disappeared in the hydronephrotic kidney. Original magnification ×240, bars = 25 µm.
PRA decreased from 15.7 ± 1.2 ng/ml/h to 10.4 ± 1.0 ng/ml/h (p < 0.05, Figure 2) after left ureteral ligation. In mice with a hydronephrotic kidney, PRA rose to 34.2 ± 3.1 ng/ml/h (p < 0.01) after losartan treatment, and 36.5 ± 2.5 ng/ml/h (p < 0.01) after enalapril treatment, respectively.
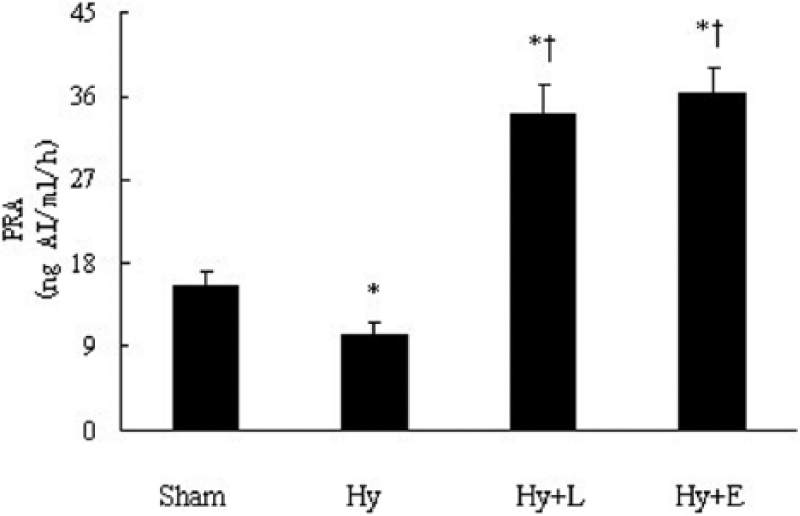
Plasma renin activity (PRA) in mice with left hydronephrosis. Sham: sham-operated; Hy: hydronephrosis; Hy + L: hydronephrosis + losartan; Hy + E: hydronephrosis + enalapril. Values are expressed as means SEM. *p < 0.05; †p < 0.01 vs Hy. n = 15 each group.
Plasma Ang I and Ang II were 15 ± 4 pg/ml and 32 ± 3 pg/ml, respectively in sham-operated animals (Table 1). Plasma Ang II level decreased in hydronephrotic mice (p < 0.05). Administration of losartan was accompanied by a rise in plasma Ang I and Ang II concentrations in hydronephrotic animals (p < 0.05). Enalapril also increased levels of Ang I (p < 0.01) and Ang II (p < 0.05) in the circulation.
ACE mRNA and protein in the heart
In mice with a left hydronephrotic kidney, cardiac ACE mRNA levels increased (p < 0.01, Figure 3(a) and (b)). After treatment with losartan, cardiac ACE mRNA levels decreased compared to the sham-operated animals (p < 0.01). Enalapril also significantly decreased cardiac ACE mRNA levels (p < 0.01). Similarly, cardiac ACE protein level was increased in mice with a hydronephrotic kidney (p < 0.05, Figure 3 (c) and (d)). Both losartan and enalapril significantly decreased ACE levels (p < 0.01), compared with sham-operated animals.

Relative cardiac angiotensin-converting enzyme (ACE) messenger RNA (mRNA) (a, b) and cardiac ACE protein levels (c, d) in sham, hydronephrotic (Hy), hydronephrotic + losartan (Hy + L)- or hydronephrotic + enalapril (Hy + E)-treated mice. Data are expressed as means SEM. **p < 0.01, †p < 0.01 vs Hy. n = 15 each group.
ACE2 mRNA and protein in the heart
In mice with the left hydronephrosis, cardiac ACE2 mRNA decreased by 25% compared with controls (p < 0.05, Figure 4(a) and (b)). However, both losartan and enalapril significantly increased cardiac ACE2 mRNA levels (p < 0.01).

Graphs showing cardiac angiotensin-converting enzyme (ACE)2 messenger RNA (mRNA) (a, b) and ACE2 protein levels (c, d) in sham, hydronephrotic (Hy), hydronephrotic + losartan (Hy +L)- or hydronephrotic + enalapril (Hy +E)-treated mice. Data are expressed as means SEM. *p < 0.05, **p < 0.01, †p < 0.01 vs Hy. n = 15 each group.
The level of cardiac ACE2 protein was decreased in hydronephrotic mice (p < 0.05, Figure 4(c) and (d)). Both losartan and enalapril significantly increased ACE2 protein levels compared with the sham-operated (p < 0.05) and with hydronephrotic mice alone (p < 0.01, Figure 4(c) and (d)).
Immunohistochemistry for ACE2 is shown in Figure 5(a–e). In the sham-operated mice, ACE2 expression was seen in cardiomyocytes (Figure 5(a)). Cardiac ACE2 immunostaining level decreased in hydronephrotic mice (Figure 5(b), p < 0.05). Losartan significantly increased cardiac ACE2 level compared with the sham-operated mice (Figure 5(c–e), p < 0.01) and hydronephrotic mice alone (p < 0.01).
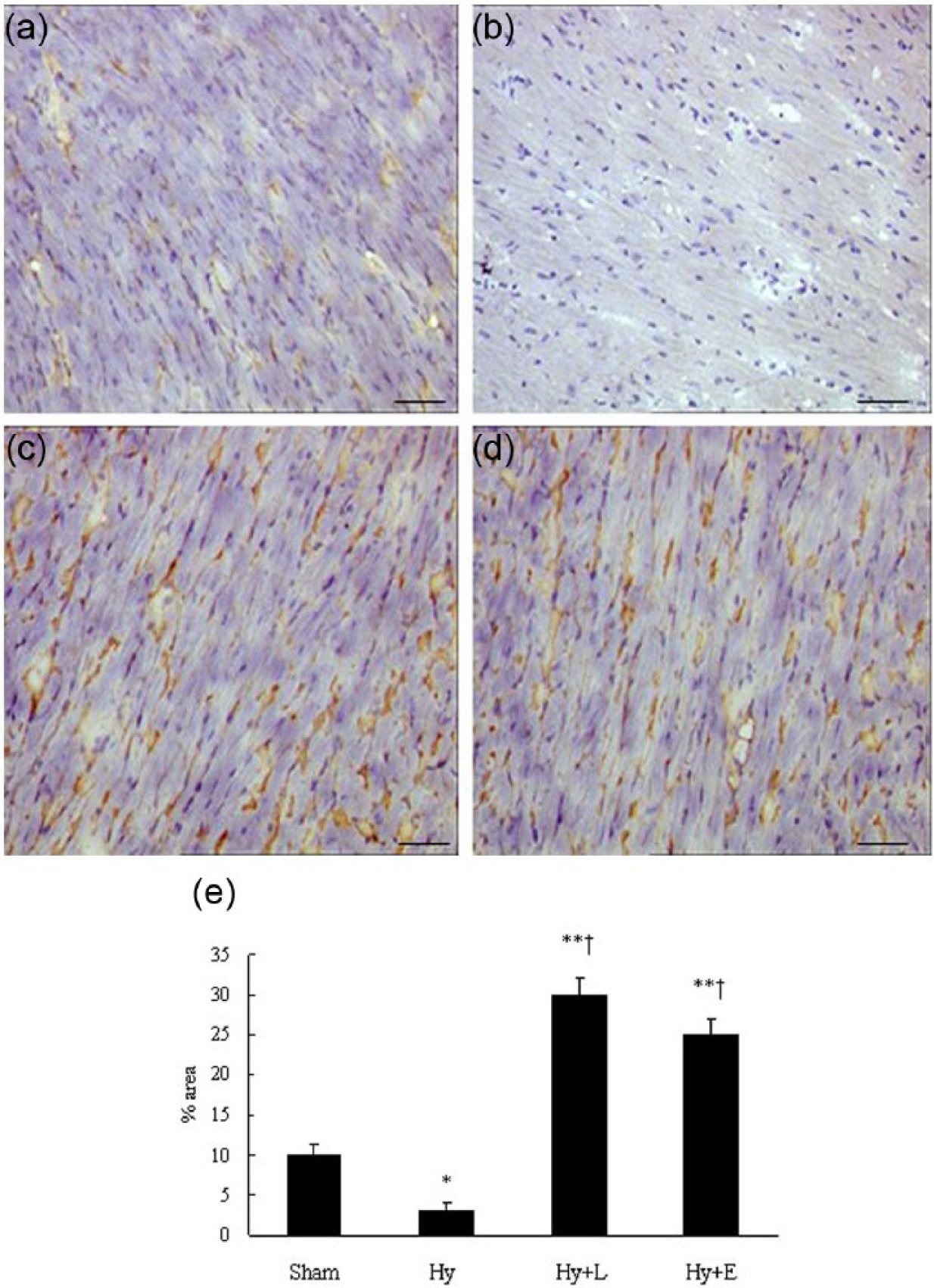
Light microscopic images of angiotensin-converting enzyme (ACE)2 immunohistochemical labeling (brown staining) of cardiomyocytes in left ventricle of sham (a), Hydronephrotic (Hy, b), hydronephrotic + losartan (Hy + L, c)- or hydronephrotic + enalapril (Hy + E, d)-treated mice. Scale bar = 100 µm; magnification, ×40. Data are expressed as means + SEM (e). *p < 0.05, **p < 0.01, †p < 0.01 vs Hy. n = 15 each group.
Mas mRNA and protein in the heart
In mice with the left hydronephrosis, the level of cardiac Mas mRNA was decreased (Figure 6(a) and (b)). Losartan treatment significantly increased Mas mRNA levels (p < 0.01) in the heart. However, enalapril had no effect on Mas mRNA expression in the hydronephrotic mice. There was a significant difference in Mas mRNA levels between the losartan- and enalapril-treated groups (p < 0.01). Furthermore, losartan significantly increased the cardiac Mas protein level (p < 0.01), but enalapril did not stimulate the cardiac Mas receptor expression (Figure 6(c) and (d)). These data suggest that the expression of cardiac Mas receptor is enhanced by losartan, but not by enalapril in this animal model.
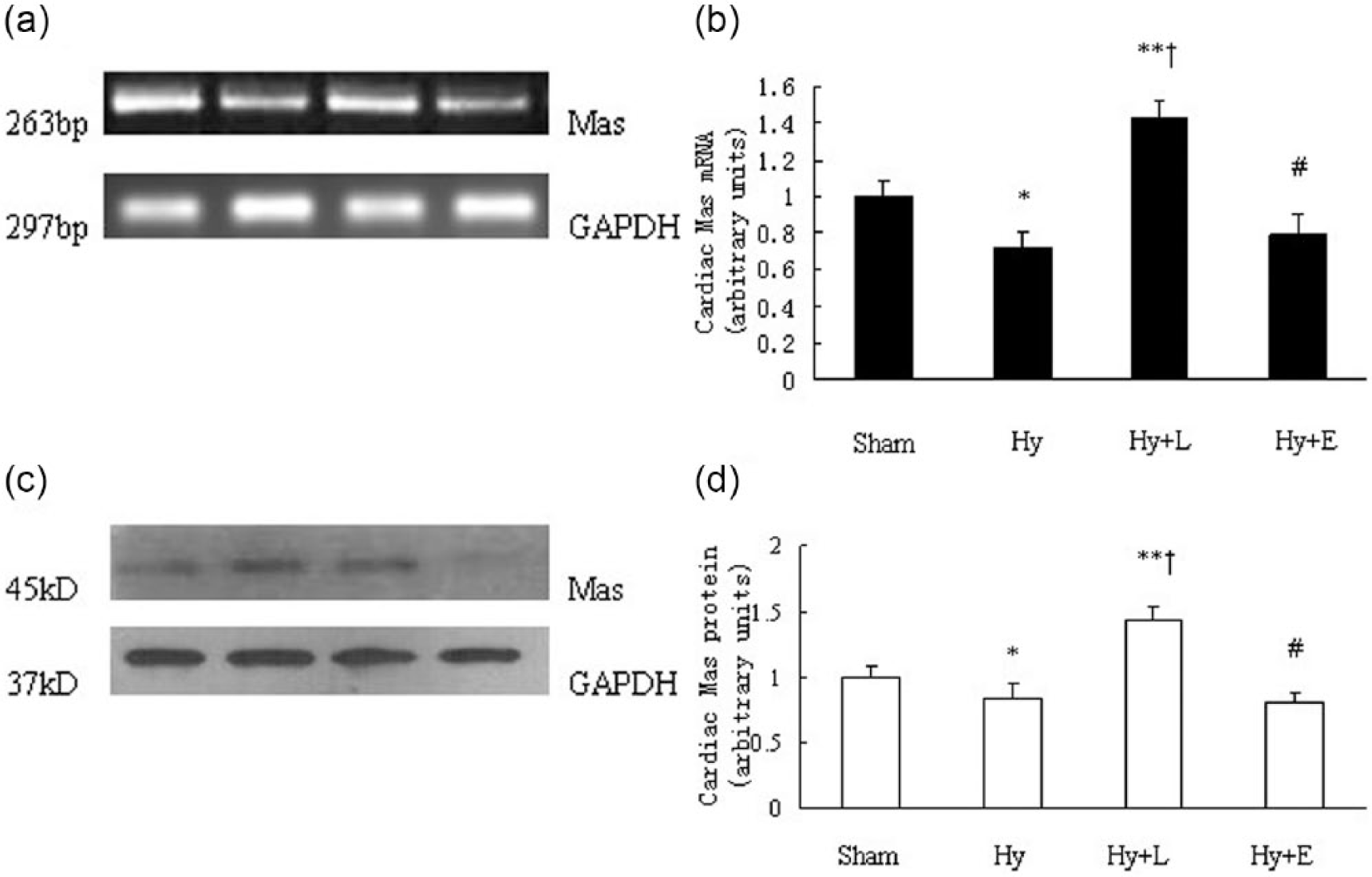
Graphs showing relative quantification of cardiac Mas messenger RNA (mRNA) (a, b) and Mas protein levels (c, d) in sham, hydronephrotic (Hy), hydronephrotic + losartan (Hy + L)- or hydronephrotic + enalapril (Hy + E)-treated mice. Data are expressed as means + SEM. *p < 0.05, **p < 0.01, †p < 0.01 vs Hy, #p < 0.01 vs Hy + L. n = 15 each group.
Discussion
Previous studies evaluated the effects of converting enzyme inhibitors and adrenoceptor blockade on renal renin secretion and synthesis in mice with a hydronephrotic kidney.12–14 This study was to determine whether hydronephrosis, caused by the left ureteral ligation, is associated with alterations in the activity of the RAS, such as levels of ACE, ACE2 and Mas receptor in the heart. This study reports for the first time that hydronephrosis can lead to an increase in the levels of cardiac ACE mRNA and its protein and a reduction in the levels of cardiac ACE2 and Mas receptor. The left hydronephrosis was accompanied by decreased PRA, indicating that renin release was less into the circulation due to the damage of the macula densa in the kidney.12–14,21 Furthermore, MAP did not significantly change in mice, suggesting that hydronephrosis in one kidney did not cause a rise in blood pressure. These results were consistent with previous reports12–14 demonstrating that the signal leading to hypertension in this model is transmitted through the baroreceptors rather than through the macula densa.2,21,22
The present study showed that both AT1 receptor blockade and ACE inhibitor decreased the levels of cardiac ACE mRNA and its protein in hydronephrotic mice. Meanwhile, the two reagents increased PRA and decreased MAP. These results demonstrated that losartan and enalapril had the effects on renin activity in the circulation and also on the RAS in the heart. Thus, we further examined whether the two reagents affect the expression of cardiac ACE2 and Mas receptor in hydronephrosis. Administration of losartan and enalapril decreased MAP and increased the levels of cardiac ACE2 in mice with a hydronephrotic kidney. However, only losartan was observed to increase the level of cardiac Mas receptor. In contrast, there was no significant effect of enalapril on the expression of the Mas receptor in these animals, suggesting that losartan and enalapril may have different mechanisms affecting the cardiac Mas receptor in hydronephrotic mice. At the present time, we do not know the exact cause of the different effects induced by losartan and enalapril on the Mas receptor; however, both reagents decreased MAP with a similar pattern, indicating that blood pressure may not be a critical factor leading to the different impacts on cardiac Mas receptor. Furthermore, similar to previous reports in rats and mice,2,12–14,21 blood pressure was not altered in mice with a hydronephrotic kidney. The normotensive status in this model suggests a pressure-independent mechanism for the alteration of Mas receptor level in hydronephrotic animals.
In previous studies, we reported that plasma renin concentration (PRC) was reduced from the hydronephrotic kidney.13,14 The present study showed that PRA and Ang II were lower in hydronephrotic mice, implying a decrease in renin activity in animals due to the damage of the macula densa, one of the critical factors controlling renin secretion.23–25 Furthermore, a level of cardiac ACE expression was elevated in hydronephrotic mice. In the present study, the changes in plasma renin are highly likely to have been associated with decreased Ang II levels, especially because of the reduction of renin release from the hydronephrotic kidney. The low levels of Ang II could stimulate the expression or activity of ACE in the heart, through a pathway of a negative regulation of Ang II on renin or ACE.26–28
In the present study, plasma Ang I and Ang II levels were elevated after administration of losartan or enalapril. Furthermore, the increase of Ang I and Ang II was accompanied by a rise of cardiac ACE2 expression in hydronephrotic animals, indicating that both Ang I and Ang II could stimulate the ACE2 levels in the heart. It is possible that ACE2 forms Ang (1–7) directly from Ang II, or through hydrolysis of Ang I to Ang (1–9) with subsequent formation of Ang (1–7).29,30
Although ACE inhibitor has similar effects of AT1 receptor blockade on cardiac ACE2 expression, only losartan increases the Mas receptor levels in the heart, suggesting that one of the main favorable effects of AT1 receptor blockade is to enhance the activity of ACE2-Ang (1–7)-Mas axis of the RAS, a protective pathway in the heart. Further work should focus on the mechanisms of alterations of cardiac RAS components observed in this study, using AT1 receptor blockers, Mas receptor antagonist and infusion of Ang (1–7). Ang (1–7) signaling and its interaction with AT1 receptor and Mas receptor signaling need to be further investigated.
These data suggest that hydronephrosis induced by the ureteral ligation causes the alteration of ACE and ACE2 expression in the heart. These results also suggest that activation of cardiac ACE2 by both enalapril and losartan may protect against the adverse effects of activated RAS and renal impairment. Thus, we propose that the change of cardiac ACE2 and Mas receptor expression induced by hydronephrosis can be an important target of strategies for preventing cardiovascular damage.
In summary, many clinical studies have discussed the renal diseases impairing cardiovascular functions. In this study, we found that hydronephrosis caused the alteration of cardiac ACE, ACE2 and Mas receptor levels. Furthermore, losartan evoked the activation both of ACE2 and Mas receptor in the heart, but enalapril had a limited effect on the Mas receptor. These findings may lead to an exciting new area in the clinical administration of AT1 receptor blockade. The observations of the different molecular mechanisms of losartan and enalapril could be helpful for better options in the treatment of cardiovascular diseases.
Footnotes
Acknowledgements
We thank B Wang for providing excellent technical assistance.
Conflict of interest
None declared.
Funding
This work was supported by the National Natural Science Foundation of China (no. 81270336).