
Abstract
Introduction:
The renin-angiotensin system (RAS) is a dynamic network that plays a critical role in blood pressure regulation and fluid and electrolyte homeostasis. Modulators of the RAS, such as angiotensin-converting enzyme (ACE) inhibitors, are widely used to treat hypertension, heart failure and myocardial infarction.
Methods:
The effect of ACE inhibitors (lisinopril and C-domain-selective LisW-S) on the constituent peptides of the RAS following myocardial infarction was examined in rats. Ten angiotensin peptides were analysed using a sensitive LC-MS/MS-based assay to examine both the circulating and equilibrium levels of these peptides.
Results:
Administration of lisinopril or LisW-S caused a significant decrease in Ang 1–8/Ang 1–10 ratios as determined by circulating and equilibrium peptide level analysis. Furthermore, Ang 1–7 levels were elevated by both ACE inhibitors, but only lisinopril decreased the Ang 1–5/Ang 1–7 ratio. This indicates LisW-S C-domain specificity as Ang 1–5 is generated by hydrolysis of Ang 1–7 by the N-domain. Further corroboration of LisW-S C-domain specificity is that only lisinopril increased the circulating levels of the N-domain ACE substrate Ac-SDKP.
Conclusion:
LisW-S is able to effectively block ACE in vivo by C-domain-selective inhibition. The LC-MS/MS-based assay allows the evaluation of the pharmacologic impact of RAS inhibitors in different pathophysiological conditions.
Keywords
Introduction
Activation of the renin-angiotensin system (RAS) plays an important role in the progression of a heart that has suffered a myocardial infarction (MI) toward heart failure. 1 Increasingly, it has become apparent that this results not only from the increased mechanical load on the heart caused by angiotensin II (Ang 1–8)-induced vasoconstriction, but also blood pressure-independent effects such as Ang 1–8-driven cardiomyocyte hypertrophy. 2 Ang 1–8 is produced from the cleavage of angiotensin I (Ang 1–10) by angiotensin-converting enzyme (ACE) and thus ACE inhibition is a prominent therapeutic approach in the treatment of post-MI patients. 3 Indeed, ACE inhibitors (ACEi) have been shown to be efficacious in treatment of MI in several large-scale clinical trials and the resultant meta-analyses.4,5 However, in 10–20% of patients the development of a persistent dry cough limits the utilisation of clinically available ACEi.6,7 Furthermore, angioedema, a life-threatening adverse reaction to ACE inhibition, is observed in 0.3% of patients. 8 It is generally accepted that these side effects are linked to the elevated levels of bradykinin induced by the concomitant reduction of bradykinin degradation by ACEi.9–11
ACE contains two homologous domains (C- and N-domain), each bearing a catalytic site, that have been demonstrated to have differential enzymatic activities. In brief, work with both N-domain and C-domain knockout mice12,13 has indicated that Ang 1–10 hydrolysis to Ang 1–8 is predominately catalysed by the C-domain whilst the antifibrotic tetrapeptide N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a highly specific substrate for the N-domain catalytic site. 14 As presently prescribed ACEi target both domains similarly, design of C-domain selective inhibitors is a logical step toward improving the side effect profile of ACE inhibition. 15 We have developed a C-domain-selective derivative of lisinopril through the substitution of a tryptophan for the P2’ proline. This derivative, LisW-S, has been shown to be highly selective for the C-domain both in vitro and ex vivo.16–18 Further to this, LisW-S has been found to be efficacious in lowering blood pressure in hypertensive mice that express active human renin in a similar fashion to that observed for lisinopril but without the increase in bradykinin levels induced by lisinopril. 19
The RAS is a complex proteolytic cascade of peptide products stemming from angiotensinogen involving multiple enzymes. Beside Ang 1–8, a variety of angiotensin metabolites have been shown to potentially influence the progression of an MI to heart failure; for example, Ang 1–7 binds to the Mas receptor, resulting in a range of cardioprotective effects 20 whilst Ang 2–8 behaves in a similar manner to that of Ang 1–8. 21 Thus, we wished to extend our understanding of the influence of LisW-S on the entire RAS in the context of MI by short-term delivery of the C-domain-selective inhibitor to normotensive rats. A recently developed liquid chromatography-mass spectrometry (LC-MS)/MS-based assay for analysing the dynamics of the constituent peptides of the RAS (termed RAS-Fingerprint22,23) was utilised to quantify plasma equilibrium levels and circulating levels of multiple angiotensin metabolites.
Methods
Induction of MI
Induction of MI was performed as previously described. 24 In brief, male Wistar rats (180–220 g) were initially anaesthetised with a mixture of oxygen and 5.0% isoflurane (Safeline Pharmaceuticals (Pty) Ltd., Johannesburg, South Africa), subsequently intubated with a 16G intravenous catheter (B. Braun Melsungen AG, Melsungen, Germany) and then placed onto a heated operating board (Braintree Scientific Inc, Braintree, MA, USA). Throughout surgery the animals were ventilated at 115 breaths/minute while anaesthesia was maintained with an oxygen/2.0% isoflurane mixture. A left thoracotomy was performed along the fourth intercostal space to expose the heart. After pericardiotomy, permanent ligation of the left anterior descending (LAD) coronary artery was performed with a 6-0 non-absorbable polypropylene ligature (Ethicon Inc, Somerville, NJ, USA) 3 mm distal the auricular appendix to induce an MI. Evidence of discolouration of the anterior ventricular wall and reduced contractility indicated a successful occlusion of the artery. For the sham operation, only a thoracotomy and pericardiotomy were performed. Thirty rats were used in this study with a 96% survival rate (n = 6–8 per group). All animal experiments were approved by the Health Science Animal Research Ethics Committee of the University of Cape Town (Ethics Committee reference number: 009/046) and were performed in accordance with the National Institutes of Health (NIH, Bethesda, MD, USA) guidelines.
Drug treatment
Osmotic pumps (Alzet, Cupertino, CA, USA) were implanted subcutaneously (as previously described 25 ) immediately post-MI. Animals were treated with lisinopril or LisW-S at 1 mg/kg per day or saline for seven days. Lisinopril was obtained from Zeneca Pharmaceuticals (Luton, UK). LisW-S was supplied by the Department of Chemistry, University of Cape Town (UCT).
Blood plasma collection
Blood was collected by cardiac puncture at the experimental endpoint. Animals were anaesthetised with halothane prior to this procedure. The following three procedures were carried out on the blood collected. For quantification of circulating angiotensin peptide metabolites, the blood was collected on ice into a tube containing an enzyme inhibitor cocktail containing ethylenediaminetetraacetic acid (EDTA), pepstatin A, p-hydroxymercuribenzoic acid, phenanthrolin and other specific RAS inhibitors (aliskiren and amastatin) to a final concentration of 5% v/v. The efficacy of the inhibitor cocktail in angiotensin peptide stabilisation was validated in whole blood. Whole blood was spiked with individual angiotensin metabolites and incubated at 37°C for 15 minutes. A greater than 90% peptide recovery following incubation indicated efficient stabilisation. For RAS equilibrium studies, blood was collected into heparin tubes and incubated for 90 minutes at 37°C prior to addition of the enzyme inhibitor cocktail to 5%. For Ac-SDKP enzyme-linked immunosorbent (EIA) assay, blood was collected on ice into a heparin tube with the ACE inhibitor captopril (final concentration 10−5 M). Blood samples were separated by centrifugation at 3000 g for 20 minutes (4°C) and plasma samples were stored at −80°C.
Infarct size analysis
Hearts were explanted, washed and perfused with saline before being transferred to a 4% paraformaldehyde solution for 24 hours before histological processing. After this fixation period, hearts were cut transversely into four equal sections and processed through graded alcohol (Illovo Sugar Ltd., South Africa) and xylene (Saarchem, South Africa) using a Tissue-Tek Rotary Tissue Processor (Sakura Finetek, Japan). Embedded in paraffin wax (Merck, Germany), the sections were cut into 2–3 μm slices, picked up on glass slides (Marienfeld GmbH & Co. KG, Germany) and baked on a hot plate at 60°C (Kunz Instruments AB, Nynäshamn, Sweden) before being dewaxed through xylene, agitated in absolute alcohol and hydrated in running tap water. Lastly, sections were stained with haematoxylin and eosin (H&E) and mounted on glass slides.
Images were acquired by microscopy (Nikon Eclipse 90i, Nikon Corporation, Japan) and captured by digital camera (Nikon DXM-1200C, Nikon Corporation, Japan).
Infarct sizes were acquired (through identification of the region lacking cardiomyocytes) and quantified with Visiopharm Integrator Systems (Visiopharm, Hørsholm, Denmark) according to the method of Takagawe et al. 26 Briefly, the sum of all midline infarct lengths was divided by the sum of all midline circumferences for all sections and expressed as a final percentage.
Ac-SDKP quantification
The concentration of Ac-SDKP in the plasma samples was quantified using an Ac-SDKP EIA assay kit (Cayman Chemicals, France). This was performed according to the manufacturer’s instructions. The 96-well microplate is precoated with a mouse anti-rabbit antibody. The assay is based on competitive binding to the coated wells between acetylcholinesterase (AChE) linked to an Ac-SDKP tracer and free Ac-SDKP in the plasma sample. Plasma samples were extracted with methanol and evaporated to dryness by vacuum centrifugation prior to assay.
Angiotensin metabolite quantification
LC-MS/MS quantification of angiotensin peptides was performed by Attoquant Diagnostics GmbH. Frozen plasma samples were shipped on dry ice to Vienna, Austria.
Plasma samples were spiked with 100 pg/ml stable isotope-labelled internal standards for each angiotensin metabolite analysed (Ang 1–10, Ang 1–8, Ang 1–7, Ang 1–5, Ang 2–8, Ang 3–8, Ang 2–10, Ang 2–7, Ang 1–9 and Ang 3–7) and 1 ml plasma was subjected to a C18 solid-phase-based sample preparation procedure (Sep-Pak, Waters, USA). Stable isotope-labelled internal standard peptides were used for normalising and monitoring endogenous angiotensin peptide recovery throughout the sample preparation procedure.
Following elution and solvent evaporation, samples were reconstituted in 50 μl 50% acetonitrile/ 0.1% formic acid and subjected to LC-MS/MS analysis using a reversed-phase analytical column (Luna C18, Phenomenex). The gradient ranged from 10% acetonitrile/0.1% formic acid to 70% acetonitrile/0.1% formic acid (t = 9 minutes). The eluent was analysed with a QTRAP-4000 mass spectrometer (AB Sciex) operated in the multiple reaction monitoring (MRM) mode using dwell times of 25 milliseconds, at a cone voltage of 4000 volts and a source temperature of 300°C. For each peptide and corresponding internal standard, two separate mass transitions were measured. Angiotensin peptide concentrations were calculated by relating endogenous peptide signals measured to internal standard signals under consideration of sample-specific response factors. A minimal signal-to-noise ratio of 10 resulted in lower limits of quantification for individual peptides of 4 pg/ml (Ang 1–10), 1 pg/ml (Ang 1–8), 2 pg/ml (Ang 1–7), 1 pg/ml (Ang 1–5), 3 pg/ml (Ang 2–8), 1 pg/ml (Ang 3–8), 5 pg/ml (Ang 2–10), 4 pg/ml (Ang 2–7), 4 pg/ml (Ang 1–9) and 1 pg/ml (Ang 3–7) .
Statistical analysis
Two-tailed student t-tests were used to assess the differences between two groups. Comparisons between multiple groups were made by means of a one-way analysis of variance (ANOVA) (Statplus-Pro, AnalystSoft Inc, USA), followed by Fisher’s least significant difference (LSD) test. Statistical significance was defined as p < 0.05. All data are expressed as means ± standard error.
Results
The hearts of the three infarcted groups were assessed with respect to size of the region that had lost perfusion after LAD coronary artery ligation. As this study was focussed on the short-term effect of ACE inhibition, hearts were explanted at Day 7 post-infarction and as such did not exhibit significant fibrosis (data not shown) and the infarction region was demarcated on the basis of the very clear loss of cardiomyocytes (Figure 1). The size of the infarct region was similar between the three groups (MI: 30.5 ± 3.7%; lisinopril: 29.7 ± 3.0%; LisW-S: 34.8 ± 1.7% p = N.S.).
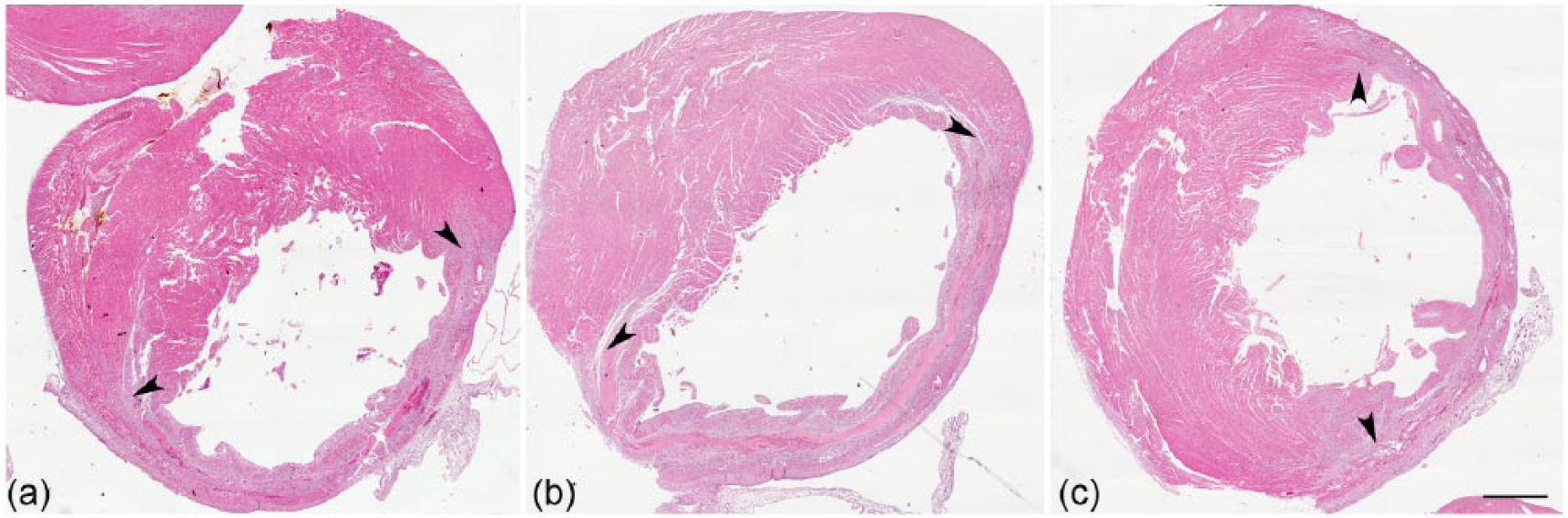
Assessment of infarct size.
Effect of LisW-S on circulating angiotensin peptide levels
Blood samples from rats were collected into tubes containing a cocktail of inhibitors that block the constitutively active peptidases of the RAS and circulating levels of angiotensin peptides were quantified using LC-MS/MS. Ang 1–8 levels were slightly increased one week following MI (by 42% p = 0.17), and a similar increase in Ang 2–8 and Ang 3–8 was observed (Figure 2). Ang 1–7 and Ang 1–5 levels did not change significantly following MI compared to sham-treated animals.

Effect of LisW-S and lisinopril on angiotensin metabolite levels in the circulating plasma at seven days post-MI.
Lisinopril administered at 1 mg/kg per day reduced the level of Ang 1–8 substantially (by 88% p < 0.01) whilst LisW-S at the same dosage resulted in a trend toward an increase in circulating Ang 1–8 (by 55% p = 0.11). However, both inhibitors caused decreases in the Ang 1–8/Ang 1–10 ratios of 21.6- and 3.5-fold for lisinopril and LisW-S, respectively, indicating inhibition of ACE-mediated conversion of Ang 1–10 to Ang 1–8. Ang 2–8 and Ang 3–8 were both seen to be substantially decreased in the lisinopril group (by 90% and 92%, respectively p < 0.01) with no significant difference observed in the LisW-S group. Ang 1–7 was not detectable in the MI group (or the sham) but was seen to increase to very similar levels in lisinopril and LisW-S (31 pg/ml). However, there were higher levels of the final breakdown product of Ang 1–7, namely Ang 1–5, in the LisW-S group relative to the lisinopril as evidenced by a 180% increase (p < 0.01).
Effect of LisW-S on whole blood equilibrium analysis
The comparison of angiotensin metabolite profiles (RAS-Fingerprints) between equilibrated whole blood from sham and MI rats revealed a slight, but non-significant, increase in equilibrium levels of Ang 1–10 and Ang 1–8, suggesting an increase in renin activity (Figure 3). The ratio between equilibrium levels of Ang II and Ang I was increased in the MI group (p < 0.05) (Figure 3 and 4(a)), suggesting an increase in ACE activity in whole blood in response to MI.

Effect of LisW-S and lisinopril on angiotensin metabolite levels in equilibrated whole blood.

The influence of LisW-S and lisinopril on ACE cleavage of Ang 1–10 and Ang 1–7 in equilibrated whole blood.
Treatment of MI rats with either lisinopril or LisW-S resulted in a dramatic increase of Ang 1–10 and Ang 2–10 relative to MI (p < 0.01 for Ang 1–10 and 2–10 with both ACEi), with the LisW-S group showing significantly higher Ang 1–10 and 2–10 levels as compared to the lisinopril-treated group. Both lisinopril and LisW-S treatment showed a strong decrease in the Ang 1–8/Ang 1–10 ratio (Figure 4), indicating efficient blockade of the C-terminal domain of ACE in whole blood of ACEi-treated animals compared to MI control rats. Equilibrium Ang 1–8, Ang 2–8 and Ang 3–8 levels were significantly reduced in lisinopril-treated animals, while remaining unchanged in the LisW-S-treated group.
Lisinopril and LisW-S caused a 10-fold and five-fold increase in levels of Ang 1–7, respectively (lisinopril: 183.4 pg/ml, LisW-S: 86.1 pg/ml; p < 0.01) compared to the untreated MI group. The ratio between Ang 1–5 and Ang 1–7, representing N-terminal ACE activity, was significantly decreased only in lisinopril-treated animals, again indicating that cleavage of Ang 1–7 was not inhibited by LisW-S.
Influence of LisW-S on levels of Ac-SDKP
Initially to determine whether LisW-S inhibited N-domain activity significantly in vivo, the levels of the N-domain specific substrate Ac-SDKP in plasma were assayed (Figure 5). As expected, lisinopril administration resulted in a highly significant three-fold increase in levels of Ac-SDKP relative to both the saline-treated sham and MI groups. No difference was observed in the level of Ac-SDKP in the LisW-S group relative to the sham and MI groups consistent with the C-domain-selectivity of the ACE inhibitor.

The differential effects of lisinopril and LisW-S on Ac-SDKP cleavage.
Discussion
In this study we have characterised the short-term response of the RAS system to a C-domain-specific ACE inhibitor in the context of MI utilising a powerful LC-MS/MS based assay. Importantly, this work demonstrates the in vivo findings C-domain-specificity of LisW-S. LisW-S was found to inhibit the catalysis of Ang 1–10 to Ang 1–8 whilst not inhibiting the degradation of Ac-SDKP, an N-domain specific substrate 14 and the conversion of Ang 1–7 to Ang 1–5.
RAS-Fingerprinting demonstrated that an MI induced an elevation in Ang 1–10 levels with a trend toward an increase in Ang 1–8, suggesting increased renin activity. This mirrors the results of Duncan et al., 27 who noted significant increases in Ang 1–10 and Ang 1–8 in infarcted rats though at an earlier time point of three days. The increase in Ang 1–10 was shown to be due to an MI-induced activation of renin. Interestingly, in contrast to previous work where there was no significant change in Ang 1–8/Ang 1–10 ratio, 27 equilibrium RAS-Fingerprint analysis elicited a slight change in this ratio, which might reflect an increase in circulating ACE activity one week after infarction.
Plasma equilibrium levels of angiotensin metabolite product-to-substrate ratios were previously shown to be suitable readouts for activities of enzymes involved in the corresponding metabolic conversions in blood or plasma sample. The RAS-Fingerprint obtained by LC-MS/MS analysis of an equilibrated sample provides an integrated picture of angiotensin-metabolizing enzyme activities present in a sample, allowing reaction-specific assessment of enzyme activities using natural, endogenously produced peptide substrates at physiologically relevant concentrations. Equilibrium angiotensin levels reflect the capability of blood or plasma to produce a certain angiotensin metabolite. The discrepancy between circulating angiotensin levels and equilibrium angiotensin levels could be explained by the lack of endothelial receptors and angiotensin-processing enzymes during the ex vivo incubation step, while renin and the high concentration of angiotensinogen is still present in the sample resulting in a constant rate of Ang 1–10 formation culminating in a stable flow-equilibrium state of peptide metabolites, which pattern is strictly dependent on the enzyme activities present in the sample. 22
Lisinopril dramatically reduced Ang 1–8, increased Ang 1–10 and correspondingly reduced the Ang 1–8/Ang 1–10 ratio in equilibrium RAS-Fingerprint analysis. This indicates that the dosage delivered is highly efficacious. However, it may also reflect the highly effective method of osmotic pump delivery as in a rat study where lisinopril administered via drinking water at 0.2 mg/kg/day did not decrease plasma Ang 1–8 whilst in the same study, where perindopril was found to be 10 times as potent as lisinopril, a dosage of 1.4 mg/kg/day of perindopril resulted in a 2.2-fold decrease in Ang 1–8 relative to the 8.8-fold reduction observed in this study. Though LisW-S did significantly inhibit ACE activity as demonstrated by the reduction in the Ang 1–8/Ang 1–10 ratios, it did not reduce the Ang 1–8 levels in the plasma. This reflects more closely the effect described above for 0.2 mg/kg/day of lisinopril and also that observed for delivery of perindopril at 0.2 mg/kg/day for 28 days after induction of an infarct in rats.28,29 This lower level of inhibition is also expected based on our previous in vitro and ex vivo results where LisW-S was found to have a lower potency than lisinopril.16,17 It is important to note that in mice where higher dosages could be achieved, LisW-S administration via an osmotic pump resulted in similar reductions in blood pressure and Ang 1–8 levels in the kidney to that seen for lisinopril. 19 The low levels of Ang 1–8 observed in the kidneys of LisW-S-treated mice are intriguing as they may in part explain the even more substantial increase in Ang 1–10 for LisW-S relative to that seen with lisinopril. The increase in Ang 1–10 is expected with ACE inhibition on the one hand due to the reduced turnover to Ang 1–8 and on the other hand due to the reduced feedback inhibition on renin release by reduced Ang 1–8/AT1R signalling. 30 The marked increase in Ang 1–10 despite a weaker systemic inhibition of C-terminal ACE in the LisW-S group suggests that ACE inhibition might be more efficacious in the kidney and/or other organs than the plasma, resulting in a stronger feedback response despite a weaker systemic effect on ACE activity.
In both the inhibitor groups, significant elevations of the cardioprotective Ang 2–1031,32 were observed as might be expected to stem from the increased levels of Ang 1–10 and the uninhibited activity of aminopeptidase (APA). A recent report further showed that APA expression is increased in murine hearts following MI, which might mediate cardioprotection via Ang 2–10, and this may be particularly beneficial at high Ang 1–10 levels. 33 Of note, in our study we did not observe an increase in circulating APA activity as indicated by the Ang 2–8/Ang 1–8 ratio in equilibrium RAS-Fingerprint analysis. However, though the effect of the inhibitors was similar with respect to Ang 2–10, lisinopril was seen to substantially reduce levels of the vasodilatory Ang 3–834,35 relative to LisW-S but also to reduce levels of the Ang 2–8, which is meant to have similar activities compared to that of Ang 1–8.36,37 The reduction of the Ang 1–8 downstream metabolites Ang 2–8 and Ang 3–8 in Lisinopril-treated animals when compared to LisW-S is most likely to be caused by the lower efficacy of LisW-S in blocking systemic ACE, as indicated by the Ang 1–8/Ang 1–10 ratio.
Both inhibitors increased the level of the strongly cardioprotective Ang 1–7 as has been observed previously for ACE inhibition. 20 It appears that neutral endopeptidase (NEP) is largely responsible for the appearance of Ang 1–7 in the circulation. 38 When the ACEi enalapril was administered to rats, Ang 1–7 was elevated but this effect was abolished by the concomitant delivery of an NEP inhibitor. Thus the increased levels of Ang 1–10 generated by ACE inhibition allow for greater traffic through NEP to Ang 1–7. Interestingly, cleavage of Ang 1–7 proceeds through ACE but there have been conflicting findings with respect to which of its two domains predominates in this reaction. The reaction has been reported to proceed via the N-domain in humans and rats39,40 but it has also been suggested that Ang 1–7 is mainly cleaved in the C-domain in canine and rat models.41,42 Our finding here shows that in vivo and ex vivo in the rat the reaction proceeds via the N-domain. Additionally it seems that the substantially elevated levels seen for Ang 1–10 in the LisW-S group still allow for circulating Ang 1–7 levels to rise comparably to the lisinopril group even though its catalysis to Ang 1–5 continues, which is supported by a similar Ang 1–5/Ang 1–7 ratio when comparing MI and MI/LisW-S groups.
Conclusion
This study shows that the C-domain inhibitor LisW-S is able to inhibit ACE in vivo in a domain-specific fashion. However, as the C-domain inhibitor appears to differentially alter levels of cardioprotective peptides (Ac-SDKP, bradykinin, 19 Ang 2–10, Ang 3–8) and peptides injurious to the heart (Ang 1–8, Ang 2–8), it is important and necessary that the ability of LisW-S to beneficially alter cardiac remodelling post-MI is determined in a long-term MI study.
Footnotes
Conflict of interest
M Poglitsch has stocks in Attoquant Diagnostics GmbH. The other authors have nothing to declare.
Funding
This work was supported by the Wellcome Trust (WT088169MA), the South African Medical Research Council (MRC) (415626), and the South African National Research Foundation (NRF) (R1008575). EDS was supported by the NRF, South Africa. NHD and PZ were supported by the MRC, South Africa.