
Abstract
Introduction:
MicroRNAs (miRNAs) are emerging as key regulators of cardiovascular development and disease; however, the cardiac miRNA target molecules are not well understood. We and others have described the Angiotensin II (AngII)-induced miR-132/212 family as novel regulators of cardiovascular function including regulation of cardiac hypertrophy, heart failure and blood pressure possibly through AT1R signalling. However, the miR-132/212 targets in the heart remain unknown.
Materials and methods:
To understand the role of these miRNAs in cardiac signalling networks, we undertook comprehensive in silico and in vitro experiments to identify miR-132/212 molecular targets in primary rat cardiac fibroblasts.
Results:
MiR-132/212 overexpression increased fibroblast cell size and mRNA arrays detected several hundred genes that were differentially expressed, including a wide panel of receptors, signalling molecules and transcription factors. Subsequent comprehensive in silico analysis identified 24 target genes, of which 22 genes were qPCR validated. We identified seven genes involved in AngII signalling pathways.
Conclusion:
We here report novel insight of an extensive network of molecular pathways that fine-tuned by miR-132/212, suggesting a role for this miRNA family as master signalling switches in cardiac fibroblasts. Our data underscore the potential for miRNA tools to manipulate a large array of molecules and thereby control biological function.
Introduction
AngII is a key hormone involved in both cardiovascular homeostasis and development of multiple cardiac diseases. 1 In the heart, sustained AngII signalling promotes myocardial hypertrophy and fibrosis through induction of several immediate early genes (c-fos, c-mys and c-jun), late genes including α-smooth muscle actin (αSMA), collagens and natriuretic peptides (ANP and BNP), and the growth factors angiotensinogen and transforming growth factor β (TGF-β).2,3 Eventually the hypertrophic and fibrotic response leads to heart failure. Hence, the AngII type 1 receptor (AT1R) is a prominent drug target in cardiovascular medicine. The classical description of AT1R signalling depicts Gαq-protein activation and downstream signalling through the canonical MAP kinases Erk1/2 that translocate to the nucleus and initiate gene transcription.4–6 We previously demonstrated that AT1R signalling also regulates five microRNAs (miRNAs), including the miR-132/212 family, in primary cultures of cardiac fibroblasts. 4
MiRNAs are endogenous short non-coding RNAs that interact with specific target mRNAs based on sequence complementarities in the 3′UTR of the target mRNA, resulting in translational repression and/or mRNA degradation.7,8 miR-132 and miR-212 are highly conserved miRNAs, closely clustered in the genome 9 and are transcribed together under the regulation of cAMP response element binding protein (CREB), 10 known to be an AngII-regulated gene. In most tissues, miR-132 is expressed at much higher levels than miR-212, which suggests that miR-132 may be the predominant regulator of the two. 11 MiRNA family members often have identical target specificity because they share common ‘seed regions’, the 2–8 bases positioned at the 5′end of the miRNA representing the primary contributors to mRNAs target recognition. However, the interactions of the 3′end of the miRNAs are also important determinants of target specificity within miRNA families. 7 Therefore miR-132 and miR-212 that belong to the same family may be functionally redundant in regulation of some target genes but not of others. 11
Several pieces of data suggest that AngII dictates miR-132 and miR-212 expression. We previously demonstrated that AngII increases miR-132 and miR-212 expression through an AT1R-Gαq-ERK1/2-dependent pathway 4 in a cell line overexpressing AT1R and in primary cultures of cardiac fibroblasts. In line with these findings, we subsequently found that miR-132 and miR-212 increased in response to AngII in hypertensive rats while these miRNAs decrease in human patients treated with AngII receptor blockers. 12 Others have shown that the miR-132/212 family promotes cardiac hypertrophy and autophagy in cardiomyocytes, 13 and that miR-132/212 null mice are protected from pressure overload-induced heart failure. 13 However, several questions regarding the mechanisms and downstream regulation of miR-132 and miR-212 targets involved in AngII signalling still remain unanswered. We therefore undertook a detailed analysis of miR-132/212 targets to understand the role of miR-132 and miR-212 in AngII signalling networks in cardiac fibroblasts.
Materials and methods
Cardiac fibroblast cell culture
Adult rat-derived fibroblasts were isolated by the principle of selective plating as previously described,14–16 with some modification. In brief, 8–10-week-old male Sprague-Dawley rats (Taconic, Denmark) were sacrificed and hearts were rinsed in a preparation buffer (1.2 mM KH2PO4, 0.25 g/l Na2CO3, 6.44 g/l NaCL, 2.6 mM KCl, 1.2 mM Mg2SO4, 11 mM glucose supplemented with 50 IE/ml Heparin). Arterial tissue and visible vasculature were removed and ventricles were trypsinised twice for 10 min in 10 ml 0.08% Trypsin (BD, New Jersey, USA) supplemented with DNase (Sigma-Aldrich, St. Louis, USA). Tissue fragments were washed, minced and incubated with 9500 U Collagenase Type 2 (Worthington Biochemical Corp., USA) in HBSS (Gibco, Life Technologies, USA) and supplemented with DNase, for 70 min disrupted by swirling. The suspension was dissociated by thorough pipetting in DMEM supplemented with 10% fetal bovine serum (FBS) and 50 U.ml-1 penicillin and 50 U.ml-1 streptomycin (PS) (Gibco, Life Technologies, USA) before being passed across a 100 µm filter (DB, New Jersey, USA). Cells were incubated for 6 min on ice with 0.9% NH4Cl, 0.08% NaHCO3, 20 µM Tetrasodium EDTA for lysis of erythrocytes and washed before the suspension was pre-plated for 1 h in cell culture dishes in DMEM/10%FBS/1%PS. After 1 h, the cultures were washed thoroughly with PBS and the remaining adherent cells were cultivated for 3 days to sub-confluence (~80%) and used directly as specified in the text. Cells were deprived of serum for 1 h prior to stimulation with 100 nM AngII (Sigma-Aldricht, St. Louis, USA).
Cell number and volume
After 44 h, cultured cells were gently detached by 0.25% Trypsin-EDTA (Invitrogen, Life Technologies, USA). Cells were resuspended and diluted in isotonic fluid and counted in the range of 8–24 µm. The cell number and cell diameter were measured using a Beckman Coulter Multisizer Z2 (RAMCON A/S, Denmark) and counting was performed in three independent experiments, each comprising triplicate measurements.
MTT assay
After 44 h cell viability was validated by the mitochondrial activity through the reduction of MTT (3-(4,5.Dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromid) by the cells, using standard Vybrant MTT proliferation assay (Invitrogen, Life Technologies, USA). In brief, cells were incubated at 37°C/5% CO2 with 0.5mg/ml MTT in phenol red-free cultivation medium for 4 h. Formazan crystals were solubilised in 10% SDS/0.01 M HCl at 80 rpm and absorbance was measured at 570–690 nm.
Immunohistochemistry
Cells were fixed for 10 min in 4% paraformaldehyde (PFA) in PBS (Bie and Berntsen LAB, Denmark), permeabilised for 10 min in 0.1% TX100 (Sigma-Aldrich, St. Louis, USA) and blocked for 15 min with 2% BSA (Calbiochem, USA) in TBS. Cells were incubated overnight in 1% BSA/TBS with mouse anti-α-smooth muscle actin (αSMA; Sigma-Aldrich; clone 1A4, 11 µg/ml), visualised by incubation for 1 h with Alexa 555-conjugated secondary antibody (Invitrogen, Life Technologies, USA: 1:200). As isotype, control mouse anti-IgG2a (Sigma-Aldrich; UPC-10) were applied. Slides were mounted with mounting medium (Vectorshield, Vector Lab, UK) containing DAPI for staining of nuclei, and images were acquired using a Leica DMI4000B Cool Fluo Package instrument equipped with a Leica DFC340 FX Digital Camera. In all experiments, exposure (camera settings) and picture processing (brief adjustment of contrast/brightness and colour balance by Photoshop CS5) were applied equally to all images
Transfection
Cardiac fibroblasts were equilibrated in serum-free medium 1 h and thereafter transfected for 4 h with 30 nM mature synthetic pre-miRNA (15 nM of each miRNA when used in combination), using Lipofectamine 2000 protocol (Invitrogen, Life Technologies, USA) as recommended by the manufacturer, except that DMEM was applied as transfection medium. Transfected cardiac fibroblasts were washed and further cultured for 24 h in serum-free DMEM. Pre-miR-132 (PM10166), pre-miR-212 (PM12534) and pre-miR scramble controls (AM17110) were purchased from Ambion, Life Technologies, USA.
mRNA and miRNA analysis
Relative qRT-PCR of mRNA and miRNA were performed as previously described.16,17 Briefly, total RNA was extracted using TriReagent protocol (Molecular Research Center, Inc. Cincinnati, USA), and RNA purity, integrity and quantity were examined by spectrophotometry (Nanodrop® Technologies, Thermo Scientific, USA) and Bioanalyzer (Agilent 2100) measurements. SYBR green-based relative quantitative mRNA PCR was performed on reverse-transcribed cDNA (High Capacity cDNA RT kit; Applied Biosystems, Life Technologies, USA) using primers listed in Table S1. For TaqMan-based miRNA, qRT-PCR primers specific for rat miR-132 (#000457), miR-212 (#002551), miR-17 (#002308), miR-191 (#002299) were purchased from Applied Biosystems, Life Technologies, USA. Amplification and detection were performed using 7900HT Fast Real-Time PCR System (Applied Biosystems, Life Technologies, USA). As recommended by others18,19 and previously described,16,17 we used the qBase+ software to normalise all qRT-PCR data against multiple stably expressed control genes (Table S2).
mRNA microarray and data processing
Total RNA samples of 500 ng were reverse-transcribed followed by in vitro transcription into biotin-labelled cRNA using the MessageAmp II Enhancer kit (Applied Biosystems, Life Technologies, USA) according to the manufacturer’s instruction for a single-round amplification. Purified and fragmented biotin-labelled cRNA was hybridised to Affymetrix® GeneChip (Rat Genome 230 2.0 Array) and subsequently stained, washed and scanned using the GeneChip Fluidics station 450 and the GeneChip Scanner 3000 (Affymetrix, Santa Clara, CA, USA). Array quality was evaluated using affyQCReport package from Bioconductor including NUSE (Normalised Unscaled Standard Error) and RLE (Relative Log Expression) plots. Normalisation and background correction was performed with R software using the vsn package by Bioconductor resulting in data that were log2 transformed. The dataset was subjected to Sylamer analysis to identify the representation of 3′UTR miRNA binding sites in genes expressed under the influence of miR-132/212 overexpression. Sylamer is a bioinformatic program that catalogues putative miRNA binding sites in the 3′UTR regions of genes and determines if the pattern deviates from neutral expectations in rank-ordered list of genes. Here, Sylamer is used to find significant depletion of a word that is complementary to the seed sequence of miR132/212, i.e. GACTGTT.
Genes identified by Sylamer analysis (~600) were subsequently analysed by Ingenuity pathway analysis (http://www.ingenuity.com) for potential targets involved in the renin–AngII signalling pathway.
Statistical analysis
All analyses comprised independent experiments, and one-way ANOVA or paired t-tests were performed as indicated (GraphPad Prism (5.0 version) software) to test significant levels. A value of p ≤ 0.05 was considered statistically significant. All error bars indicate mean ± s.d. The primary rat-derived cardiac fibroblasts were not passaged and thus the number of experiments (n) reflects the number of cell batches treated as independent experiments.
Results
AngII regulated miRNAs in vitro
We previously established an efficient protocol for isolation of primary rat-derived cardiac fibroblasts, 4 allowing us to search for potential targets of the AngII-induced miRNAs (i.e. miR-132 and miR-212) identified in our previous study.4,12 The cardiac fibroblasts were incubated with 100 nM AngII resulting in deposition of αSMA stress components, increased cell size and cell viability (Figure 1(a) and (b)). Furthermore using qRT-PCR, we confirmed that the expression of both miR-132 and miR-212 were significantly upregulated by two-fold in fibroblasts stimulated with AngII as compared with controls (Figure 1(c)).
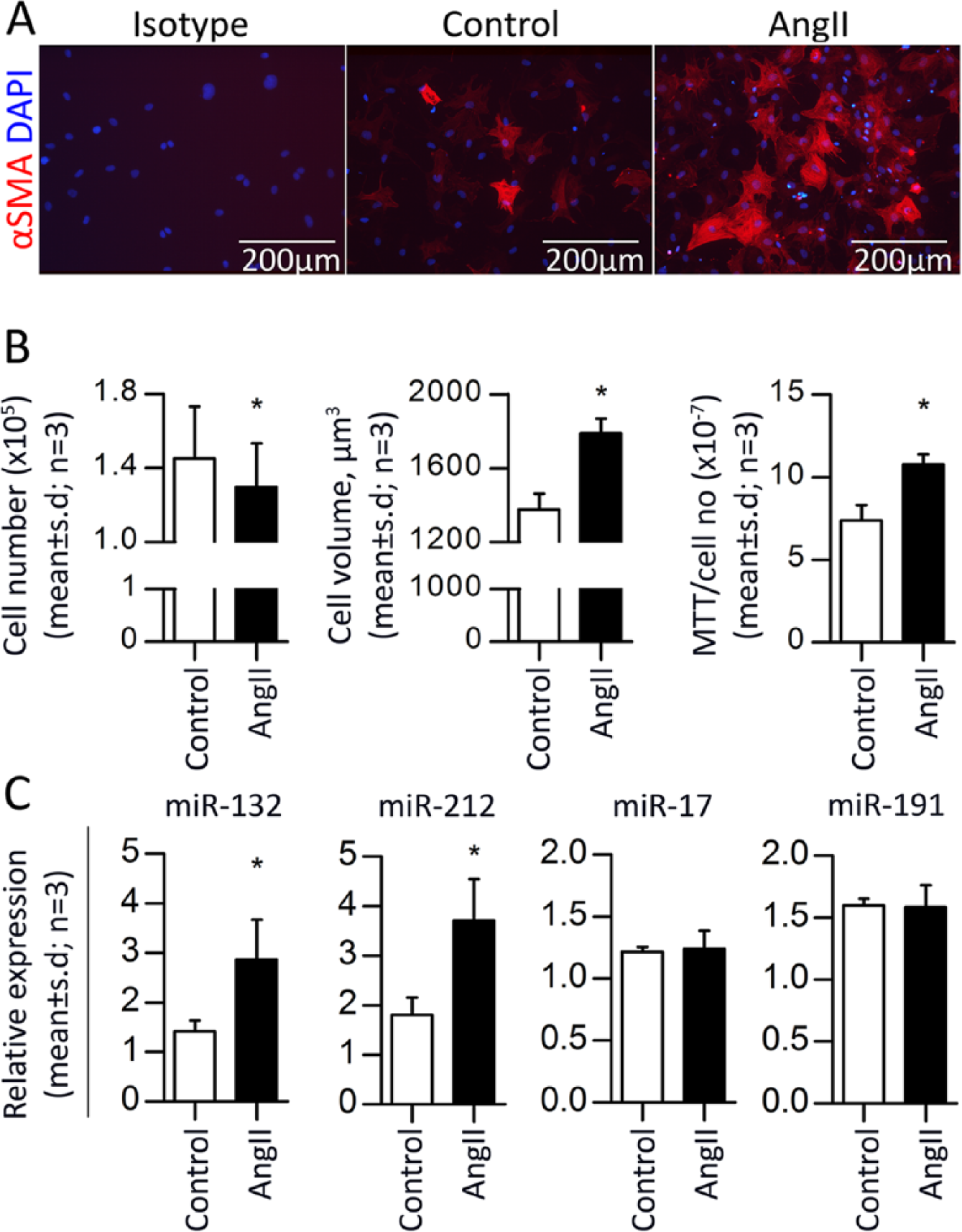
MiR-132/212 overexpression increase cardiac fibroblast size
To analyse whether miR-132/212 overexpression has an impact on cardiac fibroblasts in vitro and to search for potential miR-132/212 targets, we applied transfection of synthetic pre-miR-132 and pre-miR-212 individually (Figure S1) or in combination (Figure 2(a)). MiR-132 was increased by ~44.500 fold, whereas miR-212 was increased by ~650 fold, compared with respective pre-miR-Controls. Interestingly, we found that overexpressing a combination of miR-132/212 led to a significant (p < 0.01) cell size enlargement, indicating that miR-132/212 mimics the effect of AngII on fibroblast cell volume (Figure 2(b)) compared with controls. No changes were observed for cell number or mitochondrial activity after transfection of the miRNAs (Figure 2(b)).

MiR-132 and miR-212 target a wide panel of mRNA genes
To identify miR-132/212 molecular functions in AngII signalling pathways, we overexpressed miR-132/212 followed by a genome-wide approach identifying approximately 20,000 annotated genes, of which 2000 genes were differentially expressed (p < 0.05). Next, potential miR-132/212 targets were detected using Sylamer software and Ingenuity pathway analysis. This process identified 24 candidate target genes for the miR-132/212, of which we validated 22 genes. A flow chart of the selection process is depicted in Figure 3(a). Briefly, the software program, Sylamer, identified presence of the complementary sequence to the miR-132/212 seed region in the 33% most regulated mRNAs from the dataset (1665 downregulated and 4535 upregulated, in total 6200) (Figure 3(b)). From this analysis, 600 downregulated genes enriched for the miR-132/212 binding site were selected and compared with predicted miR-132/212 targets from three different miRNA target prediction algorithms, i.e. TargetScan, miR-base and PicTar. Interestingly, 30 genes were identified as targets by Sylamer and in at least one of the target prediction algorithms (Table S3).

Ingenuity pathway analysis of the selected 600 genes enriched for the miR-132/212 binding site identified seven miR-132/212 targets in the renin–angiotensin–aldosterone system (RAAS) (Figure 4). In general, the Ingenuity-generated dataset depicts a pattern in which more genes in the RAAS pathway are downregulated than upregulated (20/13, respectively) and several genes are identified as targets for miR-132 and miR-212 (orange circles). Furthermore, CREB is included in Figure 4, as this transcription factor is responsible for miR-132/212 transcription and because CREB has previously been suggested as a miR-132/212 target. 20

MiR-132 and miR-212 fine-tune the AngII signalling pathway
Altogether 24 predicted miR-132/212 targets were selected for validation by qRT-PCR (Table 1) together with two previous published target genes, namely p120 RasGAP (also known as RASA1; already in our list) and phosphatase and tensin homolog (PTEN). Interestingly, all target genes were found to be downregulated by overexpression of miR-132/212 (except GRB2 and PTEN). Out of 22 genes downregulated genes, two genes (DYRK2 and MAP3K3) were identified as significantly downregulated (p < 0.05) while many were borderline significant (0.05 < p < 0.2), confirming a strong tendency for these genes to be regulated by miR-132/212 in the AngII signalling pathway (Table 1). Keeping in mind that miRNAs typically exert modest inhibitory effects on many mRNAs, these results strongly support the notion that miR-132 and miR-212 function as cellular conductors fine-tuning the AngII actions in cardiac fibroblasts.
Target validation.
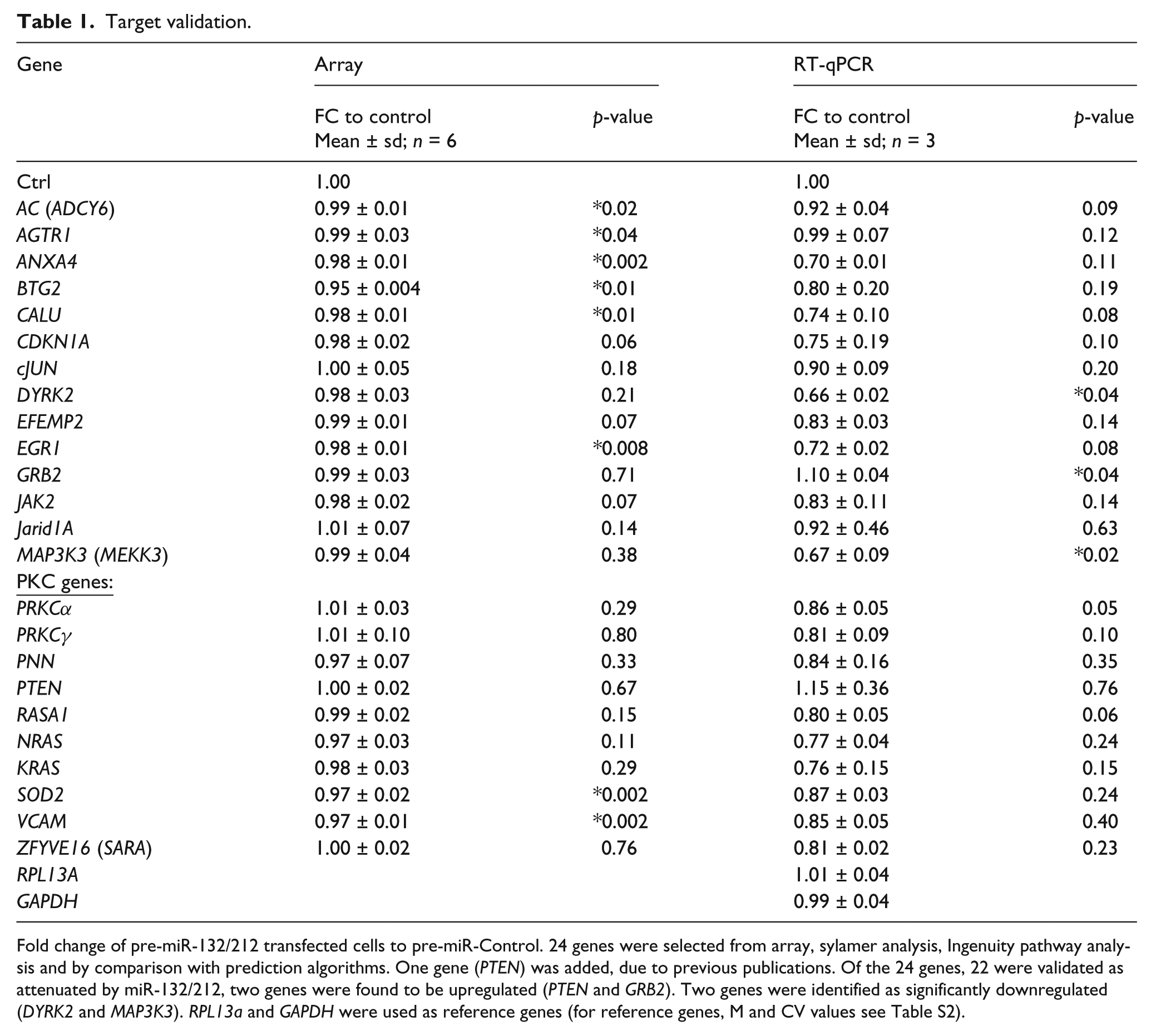
Fold change of pre-miR-132/212 transfected cells to pre-miR-Control. 24 genes were selected from array, sylamer analysis, Ingenuity pathway analysis and by comparison with prediction algorithms. One gene (PTEN) was added, due to previous publications. Of the 24 genes, 22 were validated as attenuated by miR-132/212, two genes were found to be upregulated (PTEN and GRB2). Two genes were identified as significantly downregulated (DYRK2 and MAP3K3). RPL13a and GAPDH were used as reference genes (for reference genes, M and CV values see Table S2).
Discussion
Given the emerging roles of the miR-132/212 family in cardiac hypertrophy and hypertension,12,13 it will be important to characterise the molecular networks and targets by which these miRNAs exert their functions. We previously demonstrated that signalling through the AT1R and Gαq-protein dependent pathway regulates five miRNAs, including the miR-132/212 family in HEK293N cells stably transfected with AT1R and in primary cultures of cardiac fibroblasts, but not in cardiac myocytes. 4 In this study, we identified multiple miR-132/212 targets involved in AngII, Endothelin-1 and canonical signalling pathways and suggest that the miR-132/212 family functions to fine-tune the AngII actions in cardiac fibroblasts.
Several signalling pathways activated by AngII contribute to cardiac fibroblast stress, expansion of cell volume and contribute to hypertension in vivo. Interestingly, many of the regulated targets we identified belong in these or similar pathways. Of interest, we found target genes involved in Ca2+ binding (ANXA4 and CALU),21,22 proliferation and apoptosis (BTG2 and DYRK2),23,24 endothelial to mesenchymal transition (MAP3K3 and ZFYVE16, also known as SARA),25,26 involved in cell cycle (CDKN1A, MAP3K3, NRAS, KRAS, PKC, VCAM, cJUN and RASA1) 27 as well as in AngII signalling leading to hypertrophy and fibrosis (AGTR1, AC, PKC, EGR1, JAK2, cJUN and SOD2).28-31
All genes identified as targets, except PTEN and GRB2, were attenuated in fibroblasts overexpressing miR-132/212. The increase in GRB2 might be explained by the missing miR-132/212 binding site and the fact that this molecule is activated by AT2R and subsequent Gi subunit activation30,31 and not activated by the Gq subunit accounting for most of the hypertrophic and hypertensive effects of AngII. The majority of targets were found to be attenuated borderline significantly (0.05 < p <0.2; Table 1) and two genes, DYRK2 and MAP3K3, were significantly downregulated. These results support the notion that miRNAs typically exert modest inhibitory effects on many mRNAs, which often encode proteins that govern the same biological process or gene-regulatory networks. 32
Interestingly, an evaluation of the biological effect of overexpressing miR-132/212 revealed that the fibroblast cell volume was significantly increased (p < 0.01), suggesting that overexpression of the miRNAs mimic the AngII-mediated effects. Of note, multiple factors such as serum, starvation, density and plating conditions may influence cell proliferation and cell size, 4 thus the biological relevance of the phenotypic changes observed in AngII stimulated fibroblasts may be under the influence of other factors.
Several studies identify miR-132/212 involvement in the central nervous system, i.e. in neuronal function and plasticity. From these studies, it is widely acknowledged that miR-132/212 targets a wide panel of genes including RASA1.9,10,33 MiR-132/212 has been shown to be involved in neovascularisation, targeting the endothelial RASA1. 34 In one study, miR-132 was reported to be constitutively expressed and released by pericyte progenitor cells, and transplantation of these cells into mice with myocardial infarction showed an improvement in cardiac function through proangiogenic and antifibrotic activities via inhibition of its targets RASA1 and Methyl-CpG binding protein 2 (MeCP2). 35 Moreover a recent study showed that AngII induced expression of miR-132 targets PTEN and RASA1 in vascular smooth muscle cells. 20 These targets were identified from prediction algorithms, and in vivo verification of the results was inconclusive.
It should be mentioned that in addition to miR-132 and miR-212, other miRNAs may also be involved in AngII-mediated hypertension, and likewise at the levels of mRNA targets our studies have most likely not identified all implicated mediators of the effect from miR-132 and miR-212 in AngII signalling. Also, we cannot exclude that other factors such as Endothelin-1 and TGF-β, which work synergistically to AngII, may influence the data presented herein. Another note of caution: transfection with pre-miR-132/212 led to expression levels several magnitudes higher than those elicited by AngII treatment, which may obscure comparability of the cellular effects and increase the risk of off-targets effects.
In addition to the described concept of miR-132/212 fine-tuning AngII actions in cardiac fibroblasts, it could be speculated that AngII signalling could be affected by miRNA redundancy. Thus, several miRNAs could cooperatively target various components of the signalling network or be required to sufficiently repress a single target. In addition, some miRNAs seem to ‘balance’ specific pathways by targeting both positive and negative regulatory components. We are just beginning to understand these modes of action that uphold cellular homeostasis, allowing buffering against minor physiological variations.
Genetic deletions of miRNAs in many different organisms have shown that few developmental processes are absolutely dependent on single miRNAs.36,37 Clearly, miRNA biology represents a complex mode of gene regulation.
In summary, miR-132/212 mimic and analysis of genotypic read-outs identified approximately 600 mRNAs, including 22 validated targets involved in AngII signalling. The targets were found to be attenuated and only a few significantly by miR-132/212 overexpression, supporting the notion of miR-132/212 conducting fine-tuning of AngII actions in cardiac fibroblasts.
Footnotes
Acknowledgements
We thank Tonja Lyngse Jørgensen and Charlotte Nielsen for excellent technical assistance.
Conflict of interest
None declared.
Funding
SPS is funded by the John and Birthe Meyer Foundation, TVE is funded by The Danish Cardiovascular Academy and the Danish heart foundation and DCA by The Danish National Research Council (#09-073648) and The Lundbeck Foundation.