
Abstract
Background:
IgA nephropathy is associated with increased cardiovascular risk, though whether this is due to loss of kidney function or proteinuria is unclear.
Methods:
For this study 10 normotensive IgA nephropathy subjects with early kidney disease (41±5 yrs, glomerular filtration rate (GFR) 87±9 ml/min, proteinuria 720±300 mg/d) and 10 gender- and blood pressure-matched healthy controls (36±1 yrs, estimated GFR 102±5 ml/min, proteinuria 70±6 mg/d) were studied in high-salt balance. Blood pressure and arterial stiffness, expressed as pulse wave velocity and aortic augmentation index, were measured at baseline and in response to 60 min of angiotensin II (AngII) infusion.
Results:
At baseline, IgA nephropathy subjects demonstrated similar pulse wave velocity (8.6±0.7 vs. 8.0±0.4 m/s, p=0.5) but increased aortic augmentation index (12.6±3.1 vs. 1.8±4%, p=0.04) and a trend towards increased circulating renin–angiotensin system (RAS) components (plasma renin activity, 0.55±0.18 vs. 0.21±0.05 ng/l/s, p=0.08; angiotensin II, 25±5 vs. 16±1 ng/l, p=0.08) compared with controls. However, despite similar baseline blood pressure values (p=0.8), IgA nephropathy was associated with reduced arterial sensitivity to AngII challenge (Δmean arterial pressure: 19±4 vs. 29±1 mm Hg, p=0.05; Δpulse wave velocity: -0.06±0.6 vs. 1.5±0.3 m/s, p=0.07) compared with controls, even after multivariate analysis.
Conclusion:
Even in the setting of early kidney disease, IgA nephropathy is associated with increased arterial stiffness and decreased angiotensin II responsiveness, a marker of increased RAS activity.
Introduction
IgA nephropathy (IgAN) is a leading cause of end-stage kidney disease worldwide. 1 Although progressive renal failure is a risk for cardiovascular disease, IgAN even in the absence of renal insufficiency is associated with increased cardiovascular risk 2 and increased arterial stiffness, 3 a novel and reliable intermediate risk factor for adverse cardiovascular outcomes.4,5 Though the mechanism by which IgAN with early kidney disease contributes to elevated blood pressure and arterial stiffness remains unclear, studies suggest a role for the renin–angiotensin system (RAS),6-10 up-regulation of which is known to be deleterious to cardio-renal outcomes.
We sought to clarify the role of the RAS on blood pressure and arterial stiffness modulation in the setting of IgAN with early kidney disease. We thus examined the association between IgAN with early kidney disease and blood pressure, arterial stiffness, and circulating RAS components at baseline and in response to a graded angiotensin II (AngII) challenge (3 ng/kg/min AngII × 30min; 6 ng/kg/min AngII × 30 min) as a surrogate marker for RAS activity.11,12
Methods
Subjects
Ten normotensive, non-diabetic, non-obese subjects with IgAN and early kidney disease (estimated glomerular filtration rate (GFR) >60 ml/min/1.73m2, proteinuria<1 g/d) were recruited through nephrology clinics at the University of Calgary and were enrolled after completing an initial medical history, physical examination, and laboratory screening. Medications interfering with RAS activity were stopped for 2 weeks prior to testing, a period previously shown to be adequate in terms of RAS levels returning to baseline. Gender and blood pressure-matched healthy subjects who had completed an identical protocol for a previous study were included as historical controls. All participants gave written informed consent in accordance with the Declaration of Helsinki, and the study protocol was approved by the Conjoint Health Region Ethics Board at the University of Calgary. All female subjects were studied 14 days after the first day of the last menstrual period, determined by counting days and measuring 17β-estradiol levels. 13
Protocol
Subjects consumed a high-salt diet (>150 mmol sodium/day) for 3 days before the study day to ensure maximum RAS suppression. 14 Compliance with the high-salt diet was verified by measuring sodium excretion through a 24-h urine collection or a second morning void spot urine. 15 Subjects were studied in the supine position in a temperature-controlled, quiet room after an 8-h fast.
At 8 a.m., an 18-gauge peripheral venous cannula was inserted into the antecubital vein of each arm (one for infusion, one for blood sampling). Each subject was given a loading dose of 12 mg/kg of inulin followed by a constant infusion at 30 mg/min for 90 min to establish baseline GFR measurements. After a 90 min equilibration period, blood pressure, aortic augmentation index (AIx), and brachial pulse wave velocity (bPWV) were measured at baseline and in response to a graded dose of AngII (3 ng/kg/min × 30 min and 6 ng/kg/min × 30 min). Dual measurements were repeated at 3 min intervals and values were reported as the mean of two measurements. Blood samples were collected at baseline and every 30 min until study end.
Measurement of blood pressure and arterial stiffness
Blood pressure was recorded every 15 min by an automatic recording device (Dinamap, Critikon). Subjects were studied in the supine position using a standard cuff placed on the right arm. Two readings were taken and recorded at each time point by the same registered nurse and the mean was reported. The bPWV and AIx were measured noninvasively with applanation tonometry (Millar Instruments, Houston, TX) and commercially available acquisition and analysis software (Version 8 SphygmoCor; AtCor Medical, Sydney, Australia) every 15 min, as previously described. 16 The peripheral vascular stiffness was represented by bPWV, while the central vascular stiffness was represented by AIx. We reported bPWV to evaluate the peripheral vascular response to AngII as this has been previously reported by other groups using a similar methodology.16,17 Aortic augmentation is a validated measure of central vascular stiffness and is also associated with adverse cardiovascular outcomes.16,18-21
Briefly, the AIx was determined by applanation tonometry of the right radial artery using a Millar piezoresistive pressure transducer (Miller SPT 301, Millar Instruments) coupled to a SphygmoCor device (PWV Medical). AIx was calculated from the aortic pressure waveform obtained by applying a transfer function to the radial pressure waveform. The AIx was calculated as the difference between the second systolic peak and inflection point of the aortic pressure waveform, expressed as a percentage of the aortic pulse pressure. Given that aortic pressure waveform is a combination of a forward travelling wave and a reflected wave, AIx is a parameter that reflects the degree to which aortic arterial pressure is enhanced by wave reflection. bPWV was determined by sequential acquisition of pressure waveforms from the carotid and the radial arteries by use of the same tonometer. The timing of these waveforms was compared with that of the R wave on a simultaneously recorded ECG. bPWV was determined by calculation of the difference in carotid to radial divided by the difference in R wave to waveform foot times. The distance from the sternal notch to the radial artery was used to calculate bPWV. Both these measures are validated, simple, and reproducible.18,19
Analytical methods
Plasma renin activity (PRA) and aldosterone were measured at baseline and in response to AngII infusion. An activity assay was utilized for PRA (DiaSorin Clinical Assays,Stillwater, Mn). In brief, angiotensin I, the primary product of PRA, was generated at 37°C from endogenous renin and renin substrate at pH 6.0. The integrity of the generated angiotensin I was maintained by inhibition of proteolytic activity using EDTA and phenylmethylsufonyl fluoride in the generation system. The accumulated angiotensin I reflects PRA under these controlled conditions. The angiotensin I generated was determined by radioimmunoassay (RIA) using competitive binding principles, where the antibody was immobilized onto the lower inner wall of coated tubes. Aldosterone was measured using an RIA assay. Urine total protein excretion was measured using Gen. 3 turbidimetric endpoint assay utilizing benzethonium chloride (Roche Total Protein Urine/CSF Gen. 3, Roche). Urine albumin excretion was measured using colorimetric assay utilizing bromocresol. Urine sodium was measured by an ion-selective electrode using automatically diluted specimens (ISE Indirect) and the COBAS Integra 400 (Roche Diagnostics).
Outcomes
The primary outcome was differences in blood pressure, arterial stiffness (measured as PWV and AIx), and circulating RAS components at baseline and in response AngII challenge, a well-accepted indirect measure of local intrinsic RAS activity,11,12 for patients with IgAN compared with healthy controls.
Statistical analysis
Values are presented as mean±SE unless otherwise indicated. Non-parametric methodology (Mann–Whitney U test) was utilized to compare differences between the two groups. Stepwise, backward selection multivariate regression analyses were carried out to determine the relative contribution of IgAN status on measures of blood pressure, arterial stiffness and circulating RAS components at baseline and in response to AngII challenge. The following variables were considered: age, body mass index (BMI), gender, mean arterial pressure (MAP), IgAN status (yes/no), and daily urinary protein excretion. An identical analysis was employed to determine the contribution of IgAN status on the response to AngII with the addition of baseline bPWV or AIx (as appropriate), and baseline PRA. All test assumptions were tested and verified. Statistical analyses were performed using SPSS (version 19; IBM), with two-tailed significance levels of 0.05.
Results
Baseline characteristics are presented in Table 1. IgAN subjects and healthy controls were similar in age and most were Caucasian. All subjects were non-obese, and the IgAN subjects had lower GFR (GFR 87±9 ml/min; chronic kidney disease (CKD) Stage 2) compared with healthy controls but this was not statistically significant (p=0.2). As expected, subjects with IgAN demonstrated greater proteinuria (p=0.004) and albuminuria (p=0.005) compared with healthy controls. The control subjects had both urinary protein and albumin excretion in the normal range. 22
Baseline characteristics.

Values are means± SE unless otherwise specified. BMI: body mass index; GFR: glomerular filtration rate; HbA1c: hemoglobin A1c; LDL: low density lipoprotein; HDL: high density lipoprotein; UTPE: urinary total protein excretion; UAE: urinary albumin excretion; ACEI/ARB: angiotensin-converting enzyme inhibitor/angiotensin receptor blocker.
Measured using inulin clearance technique.
Two subjects did not have biopsy done.
The majority of IgAN subjects were receiving ACEI/ARB (90%) and had received at least 6 months of steroid treatment (70%) commencing at the time of diagnosis, but none were on immunosuppressive therapy at the time of the study. Eight patients with IgAN had undergone previous kidney biopsy in which the diagnosis of IgAN was confirmed by immunofluorescence. Histologically, 62.5% of these subjects had mild mesangial proliferation (M1), 50% had mild endocapillary proliferation (E1), 62.5% had mild focal and segmental glomerulosclerosis (S1), and 75% had absent or mild tubulointerstitial fibrosis (T0). The two patients who did not undergo biopsy had the presumptive diagnosis of IgAN based on clinical presentation, as is common practice. 23 At the time of diagnosis 75% of the subjects had normal kidney function as defined by GFR, 62.5% had mild proteinuria (<1 g/day), and 100% had dysmorphic hematuria.
The unadjusted baseline blood pressure, arterial stiffness, and circulating RAS components of subjects with IgAN and healthy controls are shown in Table 2. Subjects with IgAN demonstrated greater arterial stiffness as measured by AIx after adjustment for covariates (AIx, p=0.04) but not by PWV (PWV, p=0.5). Some of the circulating measures of the RAS were greater in IgAN subjects (PRA, p=0.08; AngII, p=0.08; aldosterone, p=0.7) although not statistically significant.
Unadjusted baseline hemodynamic, arterial stiffness and circulating RAS values.

Values are means± SE unless otherwise specified. RAS: renin angiotensin system; SBP: systolic blood pressure; DBP: diastolic blood pressure; MAP: mean arterial pressure; AIx: aortic augmentation index; bPWV: brachial pulse wave velocity; PRA: plasma renin activity. *Mean of two readings.
As anticipated, there was a significant increase in measures of blood pressure, arterial stiffness and aldosterone and a decrease in PRA in response AngII challenge in all subjects (Table 3). However, IgAN subjects demonstrated a reduced blood pressure (ΔDBP, p<0.001 (Figure 1); ΔMAP, p=0.05) and arterial responses to AngII (ΔbPWV, p=0.07 (Figure 2); ΔAIx, p=0.3) compared with healthy controls. There were no differences in the PRA or serum aldosterone response to AngII challenge observed between IgAN subjects and healthy controls.
Response to angiotensin II infusion.

Values are means± SE unless otherwise specified. Δ60, value at time 60 — baseline value; SBP: systolic blood pressure; DBP: diastolic blood pressure; MAP: mean arterial pressure; AIx: aortic augmentation index; bPWV: brachial pulse wave velocity; PRA: plasma renin activity.

Diastolic pressure as function of IgA nephropathy status, at baseline and in response to AngII to AngII.
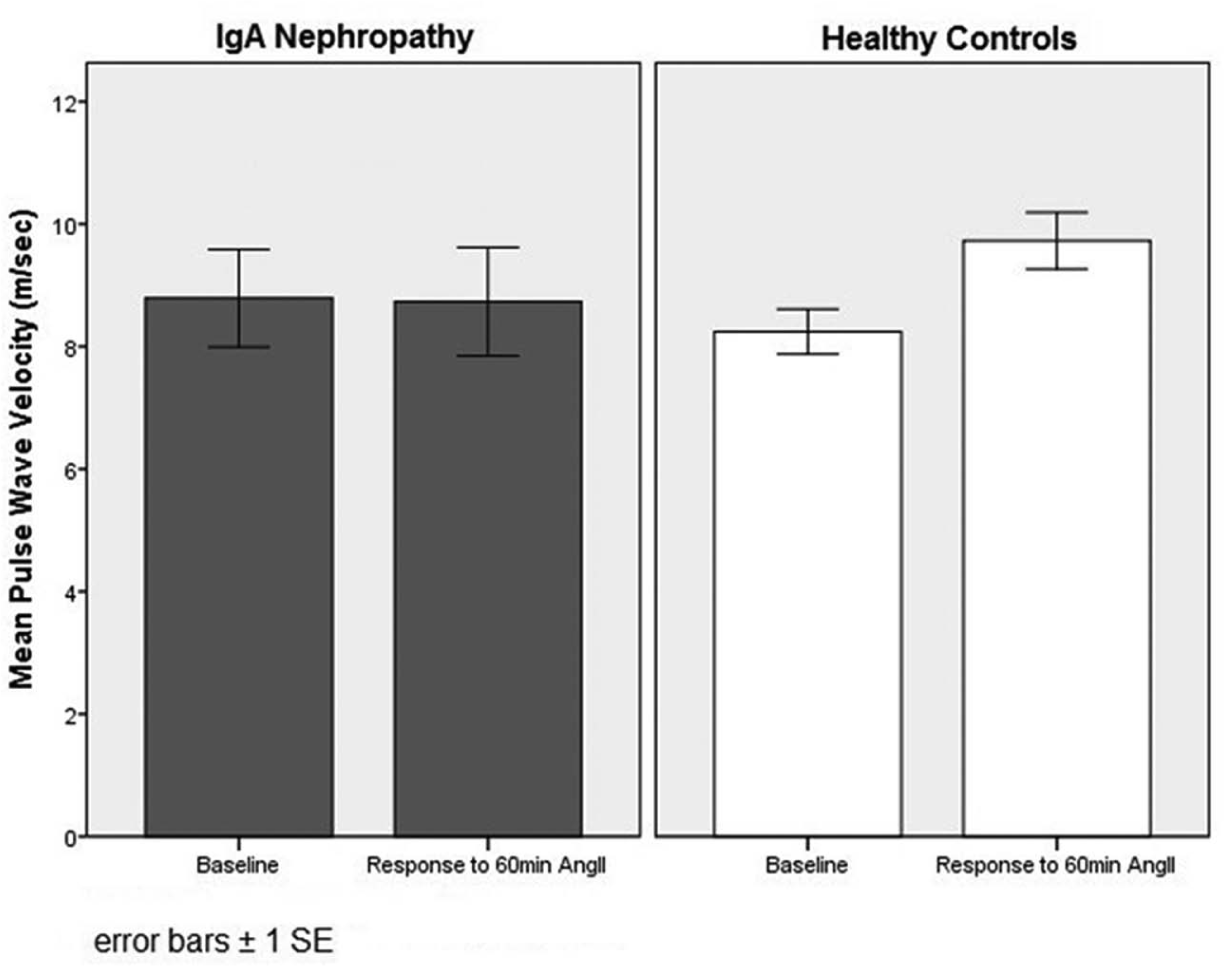
Pulse wave velocity as function of IgA nephropathy status, at baseline and in response to AngII.
Multivariate linear regression analyses revealed that IgAN status was the strongest independent predictor of baseline systolic blood pressure (SBP) (ß=0.575, p=0.015) and baseline AIx (ß=0.546, p=0.017), even after adjustment for urinary protein excretion. IgAN was also most strongly associated with most hemodynamic and arterial stiffness responses to AngII (ΔSBP: ß=1.00, p=0.027; ΔDBP: ß=0.333, p<0.001; ΔPWV: ß=-1.58, p<0.001). In contrast, urinary protein excretion was more strongly associated with baseline RAS activity (aldosterone: ß=0.650, p=0.005; AngII: ß=0.354, p=0.13) as well as renin responses to AngII infusion (ΔPRA: ß=-0.684, p=0.008). No association was observed between urinary protein excretion and other measures of circulating RAS components.
Discussion
IgAN is associated with increased cardiovascular risk 2 and increased arterial stiffness, 3 a surrogate for cardiovascular risk,4,5 even in the absence of conventional cardiovascular risk factors.24,25 To our knowledge, this is the first study examining the relationship between IgAN with early kidney disease and AngII-dependent control of blood pressure and arterial stiffness in humans under controlled dietary conditions. Our key findings were as follows: compared with healthy controls, IgAN status with early kidney disease was independently associated with a) increased arterial stiffness and a trend toward elevated circulating RAS components and b) decreased arterial sensitivity to AngII challenge, as demonstrated by the blunted blood pressure and arterial vasoconstrictor response to exogenous AngII.
The implications of our findings are best understood in the context of prior studies using a similar methodology.26-30 Shoback et al. originally hypothesized that a blunted vascular response to AngII was a reflection of high local tissue AngII concentrations and tissue renin–angiotensin-aldosterone system activity. 28 Our findings of a decreased sensitivity to AngII in subjects with IgAN compared with gender and blood pressure-matched controls thus, considering this hypothesis, likely reflects differences in tissue-specific RAS activity. 31
Previous studies have reported increased arterial stiffness in patients with CKD, 32 though few have studied patients with specific etiologies of CKD or investigated potential underlying mechanisms. Kesoi et al. demonstrated that IgAN patients with Class 1–4 CKD have increased arterial stiffness as measured by ambulatory arterial stiffness index, 3 though we are unaware of any studies specifically examining arterial stiffness as measured by PWV or AIx in patients with IgAN, particularly those with early kidney disease.
Limited studies link IgAN to increased RAS activity.6,7 A growing body of evidence suggests that therapeutic blockade of the RAS in IgAN is associated with improved outcomes.8-10 In support of the present findings, Lai et al. demonstrated reduced mesangial cell expression of angiotensin type 1 receptor after acute exposure to polymeric IgA1 in IgAN patients as a function of negative feedback due to increased intraglomerular RAS activity. 33 However, this adaptation is gradually lost with prolonged exposure to polymeric IgA1, offering a potential explanation for the eventual deterioration in kidney function observed in patients with IgAN, particularly those with proteinuria. 34 The increased circulating levels of RAS components and blunted arterial response to AngII challenge observed in the IgAN subjects in our study are in keeping with these and other reports. 35 These findings suggest that RAS interruption may optimize cardiovascular outcomes in this population, as has been demonstrated in other high-risk populations,36,37 with pharmacological RAS blockade decreasing measures of arterial stiffness both acutely and chronically (>4 weeks). 38
Our study has strengths and limitations. First, our small study sample may have limited our ability to detect small differences between the groups. Our study was also limited to IgAN subjects with normal blood pressure, mild loss of kidney function and <1 g/d proteinuria. However, by studying a healthy, homogeneous population and employing a careful pre-study design, we aimed to examine the association between IgAN and measures of arterial stiffness by minimizing confounding factors. We ensured that all participants were in high-salt balance to ensure maximum RAS suppression and that all female subjects were studied during the same stage of the menstrual cycle and not ingesting oral contraceptive, to control for the effect of estrogen on the vasculature and the RAS. 13,29 Proteinuria is associated with increased RAS activity, both in this study and in others.39,40 However, even after controlling for proteinuria, IgAN remained an independent predictor of arterial measures, both at baseline and in response to AngII. As it is not possible to measure vascular RAS activity directly in humans, we used a surrogate marker of measuring RAS in humans by observing the response to Ang II challenge, as has been done by others.11,12 Though medications that interfere with the RAS were discontinued for 2 weeks prior to the study to allow RAS activity to return to baseline levels, it remains possible that long-term effects of drug therapy on arterial physiology might still be relevant. Finally, due to the cross-sectional nature of our study design, we cannot comment on causality. Nevertheless, our study suggests that IgAN with early kidney disease is independently associated with increased blood pressure and arterial stiffness as well as decreased arterial sensitivity in response to AngII challenge compared with healthy controls. Larger, prospective studies are required to determine pathophysiological mechanisms of cardiovascular disease progression in the setting of IgAN with early kidney disease and significant proteinuria.
Summary
IgAN is associated with increased cardiovascular risk and increased arterial stiffness, a surrogate for cardiovascular risk, even in the absence of conventional cardiovascular risk factors. Our study is the first study examining the relationship between IgAN with early kidney disease and AngII-dependent control of blood pressure and arterial stiffness in humans under controlled dietary conditions. It suggests that IgAN with early kidney disease is independently associated with increased arterial stiffness as well as decreased arterial sensitivity in response to AngII challenge compared with gender and blood pressure-matched healthy controls.
Footnotes
Acknowledgements
An abstract for this study was presented at the annual meeting of the American Society of Nephrology in San Diego, CA, October 31–November 4 2012.
Author contributions
A.A.A., M.C.M., B.R.H., J.M.M., T.T.C., B.H., D.Y.S., and S.B.A. provided conception and design of research; A.A.A., M.M., and S.B.A. analyzed data; A.A.A., M.C.M., and S.B.A. interpreted results of experiments; B.H. interpreted the biopsy results; A.A.A. and M.C.M. prepared figures; A.A.A. and S.B.A. drafted manuscript; A.A.A., M.C.M., B.R.H., J.M.M., T.T.C., B.H., D.Y.S., and S.B.A. edited and revised manuscript; A.A.A., M.C.M., B.R.H., J.M.M., D.A.M., T.T.C., B.H., D.Y.S., and S.B.A. approved final version of manuscript; D.Y.S. and S.B.A. performed experiments.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by an Establishment Grant from Alberta Innovates-Health Solutions. M.C. Mann is supported by a Canadian Institutes of Health Research doctoral award. B. R. Hemmelgarn, and S. B. Ahmed are supported by Alberta Innovates-Health Solutions and the Canadian Institute of Health Research. B. R. Hemmelgarn, J.M. MacRae and S. B. Ahmed are supported by a joint initiative between Alberta Health and Wellness and the Universities of Alberta and Calgary. Funding sources had no role in the design, conduct, or reporting of this study.