
Abstract
Apart from the well-documented role of the renin–angiotensin–aldosterone system (RAAS) in regulating the blood pressure and other related parameters, its role in modulating different physiological/pathological functions, including pain, has also been described. Like its dual role in regulating stress-related anxiety and cognitive functions, its dual role has also been documented in pain modulation in different disease states. Drugs blocking the RAAS activation, viz., renin inhibitors, angiotensin converting enzyme (ACE) inhibitors, AT1 receptor antagonists and aldosterone antagonists, have been shown to produce beneficial effects in migraine and neuropathic and nociceptive pain. Their beneficial effects have been mainly attributed to inhibition of the inflammatory cascade of reactions by inhibiting the generation of key cytokines, including tumor necrosis factor (TNF)-α. On the contrary, clinical as well as preclinical studies have also shown the pain-inducing actions of renin–angiotensin system (RAS) blocking drugs. Furthermore, the pain-relieving actions of angiotensin II (AngII) and pain-inducing actions of AT1 blockers have also been described. The pain-inducing actions of ACE inhibitors have been mainly attributed to interference with metabolism of bradykinin and substance P, while the analgesic actions of AngII have been mainly related to activation of brain localized AT2 receptors and release of endogenous opioids. The present review describes the dual role of the RAAS in different states of pain.
1. Introduction
The renin–angiotensin–aldosterone system (RAAS) is an important regulator of the blood pressure and the fluid/electrolyte homeostasis. 1 However, apart from its very well-documented cardiovascular effects, its role in modulating different physiological/pathological functions has also been described. The components of the renin–angiotensin system (RAS) present in the central nervous system modulate sensory information, emotional and behavioral responses, stress, anxiety, learning and memory.2,3 The angiotensin neuropeptides have been shown to impair the cognitive functions and, accordingly, angiotensin inhibitors improve learning and memory.4–6 In contrast, studies from our laboratory and from others have described the learning-facilitating and memory-enhancing effects of angiotensin neuropeptides.7,8 Furthermore, evidence from pharmacological studies suggests the dual role of RAAS in modulating stress and associated anxiety in different models of stress.9–11 These effects have been mainly attributed to differential changes in AT1 and AT2 receptor expression in the stress-responsive brain areas, both inside and outside the blood–brain barrier.12–14
The AT1 and AT2 receptor subtypes are widely distributed in different brain areas, including the key areas controlling nociception, such as the anterior cingulated cortex (ACC), prefrontal cortex (PFC), thalamus, periaqueductal gray matter (PAG), amygdala, nucleus accumbens and spinal cord.15–17 The PAG of the mid-brain region plays an important role in descending pain modulation by activating enkephalin-releasing neurons projecting to the raphe nuclei in the brain-stem. 18 The amygdala, located deep within the medial temporal lobes of the brain, is an important component in an emotional–affective dimension of pain, including neuropathic pain. The amygdala includes several nuclei, including the lateral (LA), basolateral (BLA) and central (CeA) nuclei, for sensory and pain processing. 19 The ACC is the forebrain structure that plays an important role in regulating the affective and emotional component of physiological as well as pathological pain.20–22 The orbitofrontal cortical area of the PFC is an important component of a flexible cerebral network involved in acute and chronic pain processing and perception. 23 The thalamus is the major relay between a variety of subcortical areas and the cerebral cortex, and its key role in chronic pain has been reported in both clinical and animal experimental studies. 24 The spinal cord is the major conduit for sensory (pain) information from the periphery to the brain region, and a series of molecular changes in spinal cord and brain centers are associated with central pain sensitization in neuropathic pain. 25
A number of clinical as well as preclinical studies have identified the role of the RAAS in diverse states of pain (Table 1). The drugs blocking the activation of the RAAS have been shown to produce beneficial effects in migraine and in neuropathic and nociceptivepain.26–28 In contrast, there has also been evidence showing the pro-nociceptive effects of RAS-blocking drugs.29,30 The present review describes the dual role of RAAS in different states of pain.
Pain attenuation and pain-inducing effects of various interventions in different models of pain.

ACE: angiotensin converting enzyme; CCI: chronic constriction injury; CRPS: complex regional pain syndrome.
2. Pain-inducing role of RAAS
2.1 Analgesic profile of drugs blocking RAAS
2.1.1 Preclinical studies
The evidence regarding the pain-inducing role of the RAS in the body has mainly come from studies employing different pharmacological blockers in experimental pain models. Marques-Lopes and co-workers demonstrated that exogenous microinjection of AngII in the spinally-projecting caudal ventrolateral medulla (CVLM) elicits hyperalgesic effects (assessed by the tail-flick and formalin tests), which were attenuated by local administration of the AT1 antagonist, losartan. The involvement of the RAS in pain modulation in the supra-spinal areas is mainly due to AT1-expressing CVLM neurons projecting to the spinal cord. 31 The same group of scientists demonstrated that AngII-induced hyperalgesia, elicited from the CVLM, is mediated to the spinal cord by an indirect pathway relayed at the pontine noradrenergic A(5)group. 32 A very recent study has demonstrated that the systemic continuous delivery of sub-pressor doses of AngII (150 ng/kg/min) induces pain in terms of tactile and cold hyperalgesia in chronic constriction injury (CCI)-subjected animals which was shown to be attenuated with losartan. Furthermore, a marked increase in large-sized injured primary afferent neurons was detected on the ipsilateral side after AngII treatment, suggesting that AT1 receptor activation is an important regulatory factor in neuropathic pain perception. 33
Studies from our laboratory have shown the beneficial effects of RAAS blockers in experimental models of neuropathic pain. Administration of different doses of aliskerin (a direct renin inhibitor), telmisartan (AT1 receptor antagonist with highest affinity) and spironolactone (aldosterone receptor antagonist) for 14 days attenuated neuropathic pain in terms of decrease in paw cold allodynia, mechanical allodynia and heat hyperalgesia in CCI-induced neuropathic pain in rats.26,27,34A study from another laboratory has also demonstrated the dose-dependent anti-nociceptive actions of systemically administered aliskerin in the writhing, formalin hindpaw, capsaicin-induced pain, post-operative pain, CCI-induced neuropathic pain and orofacial pain tests in mice. 35 A study from our laboratory has also reported the anti-hyperalgesic effect of spironolactone in diabetic neuropathic pain. 28 Spironolactone has also been shown to attenuate acetic acid-induced inflammatory visceral pain in mice. 36 The repeated administration (not single dosing) of spirapril and losartan has been shown to elicit anti-nociceptive actions, suggesting the nociceptive role of the brain endogenous AngII. 37
2.2.2 Clinical studies
Though migraine is not generally considered as neuropathic pain, some studies have drawn similarities between migraine and neuropathic pain and, hence, migraine may be classified as a type of neuropathic pain. 38 Clinical studies have shown the important role of RAS in migraine pathophysiology 39 and, thus, AT1 receptor blockers and ACE inhibitors have potential in the prophylactic management of migraine. ACE inhibitors (including enalapril and lisinopril) and angiotensin AT1 receptor blockers (including telmisartan) are shown to be effective in preventing attacks of migraine40–44 (Figure 1).

Analgesic and hyperalgesic action of drugs blocking the renin–angiotensin–aldosterone system.
2.2 Possible mechanisms
In our studies, administration of aliskerin, telmisartan and spironolactone was shown to attenuate CCI-induced rise in TNF-α level in the sciatic nerve. The role of TNF-α and other cytokines in development of peripheral as well as central sensitization in neuropathic pain is very well documented.45,46 The up-regulation of TNF-α has been detected at the injury site, mainly in the macrophages and Schwann cells.47,48 Nerve biopsies from patients with painful neuropathy also show higher levels of TNF-α expression, especially in the Schwann cells. 49 The bilateral elevation of TNF-α has also been documented in the lumbar and cervical dorsal root ganglia after unilateral chronic constriction injury of the sciatic nerve. 50 An intra-sciatic injection of TNF-α has been shown to produce pain hypersensitivity similar to that of neuropathic pain in humans, 51 and neutralizing antibodies to TNF-α receptors have been documented to attenuate pain hypersensitivity. 52 It has been documented that the up-regulation of TNF-α in the peripheral nerve region is followed by an increase in the cytokine levels in the neurons of dorsal root ganglia of the spinal cord. 53
The AT1 receptors are localized on the different inflammatory cells54,55 and the critical role of RAS activation in various processes of inflammation including accumulation of neutrophils, 56 differentiation of dendritic cells 57 and production of inflammatory chemokines has been documented. 57 Studies have also documented the potent anti-inflammatory potential of aliskerin by inhibiting leukocyte adhesion, preventing oxidative stress and decreasing the production of inflammatory cytokines, including interleukin(IL)-6 and TNF-α.35,58–60 Furthermore, aliskerin has also been shown to attenuate TNF-α-mediated deleterious effects on the different body systems. 60 Aliskerin-induced decrease in inflammatory mediators may possibly be attributed to decrease in AngII levels. The molecular mechanisms involved in AngII-induced development of inflammation and, consequently, pain are depicted in Figure 2. Studies have identified the beneficial effects of spironolactone in various disease states mainly due to its ability to inhibit the production of different pro-inflammatory cytokines. 61 Spironolactone has been reported to modulate retinal inflammation, with potential to treat retinal vasculopathy. 62 Treatment with spironolactone has been shown to improve the endothelial dysfunction and inflammatory disease activity in rheumatoid arthritis. 63

Molecular involvement of angiotensin during pain signaling.
Although the beneficial effects of spironolactone in attenuating neuropathic pain have been described as being due to its aldosterone receptor blockade activity, some studies have suggested that the suppressive effect of spironolactone on immune-reactive and inflammatory cytokines is independent of mineralocorticoid receptor blockade. Furthermore, the direct role of pro-renin receptors in decreasing inflammatory cytokines and producing beneficial effects in neuropathic pain may not be completely ruled out in aliskerin-mediated beneficial effects. 64 Apart from the indirect role of renin in inflammation through AngII formation, its direct effects have also been demonstrated through the (pro)renin receptor (PRR), which is in fact a receptor for both renin and its precursor, prorenin. Both renin and prorenin bind to PRR to initiate a complex cascade of inflammatory signaling pathways in an AngII-independent manner.65,66 PRP-triggered intracellular signaling includes mitogen-activated protein kinases (MAPK), including extracellular signal-regulated kinase (ERK1/2), p38-heatshock protein 27 and PI3K-p85 pathways. In response to PRR-triggered signaling, there is an up-regulation of transforming growth factor (TGF)β1, cycloxygenase-2, TNF-α, IL-1β, VEGF and ICAM[AU: please define VEGF and ICAM] that may initiate and amplify the inflammatory cascade.67,68 The role of the above-described signaling pathway including MAPK has been very well described in the development of different types of pain, including neuropathic.69,70
Similarly, aldosterone may also bind to various receptors’ channels to initiate an inflammatory signaling cascade in an AngII-independent manner. The binding of aldosterone to its cytosolic mineralocorticoid receptor may initiate genomic as well as non-genomic actions. The latter actions are fast in onset and include activation of protein kinase C (PKC), protein kinase D (PKD) and MAP kinase cascade including ERK1/2.71,72 Aldosterone-induced activation of these signaling cascades is actually a consequence of transactivation of the epidermal growth factor receptor (EGFR) via the non-receptor tyrosine kinase, c-Src. 72 The non-genomic actions of aldosterone may also involve production of reactive oxygen species, activation of NF-κB and Rho/Rock2kinases.73,74 The role of the above-described signaling cascade including Rho/Rock2 kinases has been well described in different types of pain.70,75,76 The inflammatory action of aldosterone may also involve activation of transient receptor potential melastatin 7 cation channels (TRPMC) through mineralocorticoid receptors, 77 whose role in pain development is also described. 78 Furthermore, the rapid response of aldosterone has also been linked to activation of G protein coupled estrogen receptors (GPER), independently of mineralocorticoid receptors. 79
Based on this, it may be proposed that the beneficial effects of RAAS blockers in experimental models of neuropathic pain are mediated indirectly through their attenuating effect on pro-inflammatory mediators. AT1 receptor blockers-induced inhibition of neurogenic inflammation may also be responsible for their beneficial effects in migraine.80,81 The CCI model in rats mimics the complex regional pain syndrome in humans,82,83 which in turn is characterized by the activation of the sympathetic nervous system. Therefore, it may be proposed that nerve injury induces the activation of the sympathetic nervous system, leading to activation of the RAAS with a consequent increase in formation/release of renin, angiotensin and aldosterone, leading to initiation and maintenance of neuropathic pain43,49 (Figure 3).
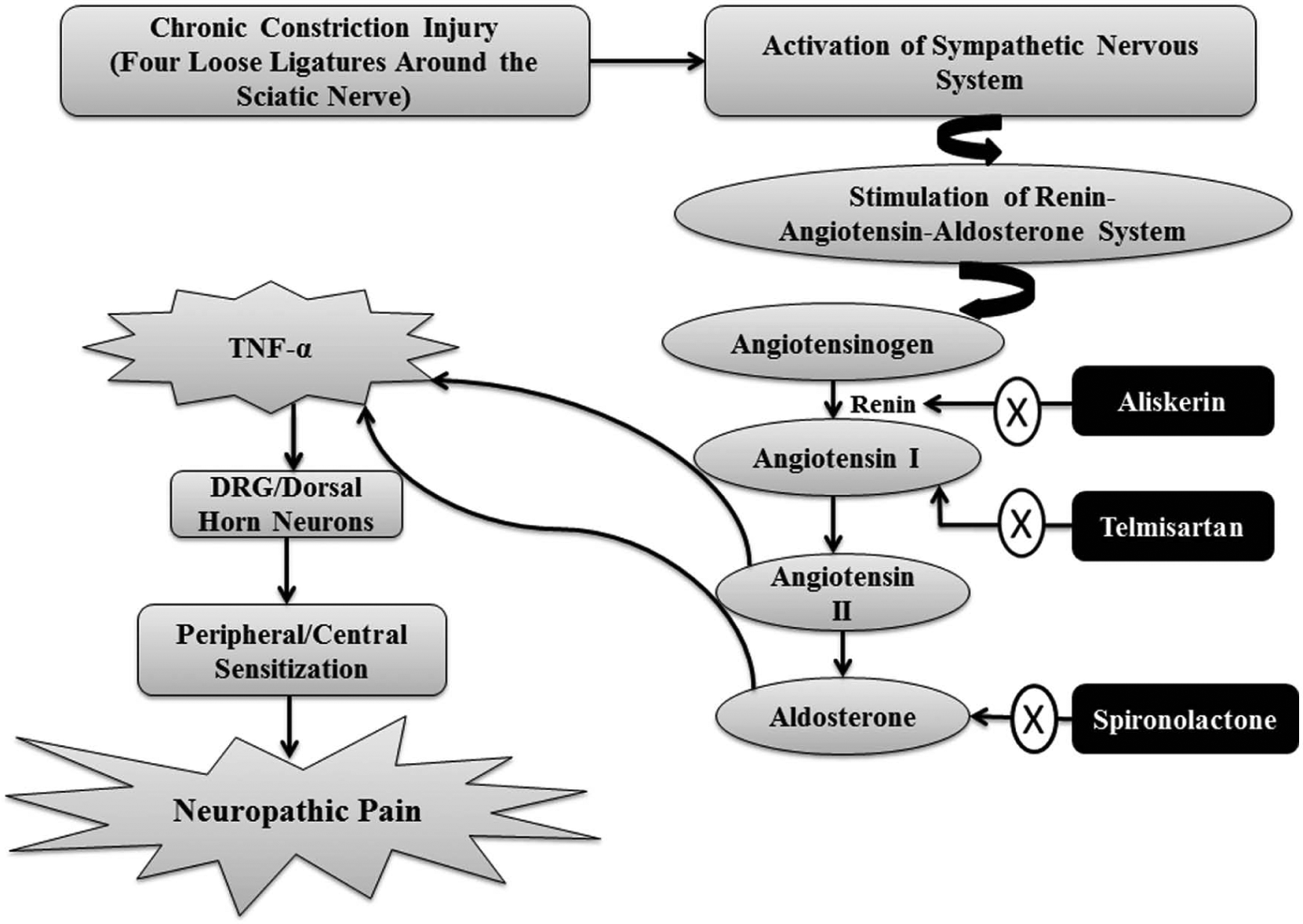
Possible hypothesis regarding the role of the renin–angiotensin–aldosterone system in development of neuropathic pain in chronic constriction injury-subjected rats.
3. Pain-attenuating effects of RAAS
3.1 Preclinical studies
There have been a number of preclinical studies showing the anti-nociceptive actions of exogenously administered AngII in various pain models.15,84,85 Georgieva and Georgiev demonstrated the dose-dependent anti-nociceptive effects of intracerebroventricularly (ICV) administered AngII (0.05, 0.1, and 1 mg) in the acetic acid (1% i.p.)-induced writhing model in male albino mice. 15 The microinjection of AngII into the ventrolateral PAG (the gray matter located around the cerebral aqueduct within the tegmentum of the midbrain) has been shown to exert anti-nociceptive effects in the tail flick test and incision allodynia. 19 ICV injection of AngII (10 and 20 ng) has also been shown to increase the tail flick latency in rats. 86 Intra-cerebral administration of renin and AngII has also been shown to increase the latencies to thermoalgesic stimuli in animals. 84 The dose-dependent anti-nociceptive effects due to microinjection of porcine renin substrate tetradecapeptide and different angiotensin peptides including AngII into the ventrolateral PAG have also been described in the rat tail flick test. 85
It is well documented that spontaneous hypertensive rats (SHR) exhibit decreased pain sensitivity, which may be possibly ascribed to over-activation of the RAS in the body.87–89 Irvine and White demonstrated that peripheral administration of AngII (567 mg/kg/day) to normal rats reduces the pain sensitivity (assessed in terms of increased hot plate latencies) equivalent to that observed in SHR. In contrast, chronic ICV administration of AngII (14 µg/kg/day) in normal rats did not alter the pain threshold in a significant manner. Furthermore, ICV administration of losartan to SHRs did not produce any effect on nociception, suggesting that the increased levels of AngII in the periphery are mainly responsible for reduced pain perception inSHR. 90 The preclinical studies showing an increase in pain perception (in terms of increase in the number of writhings) in the thermal and chemical nociception model of pain with captopril and losartan also suggests the pain-attenuating effects of endogenous AngII. 81
An active role of the endogenous RAS in producing analgesia is further supported by stress-induced analgesia-based studies.84,86 Studies have shown a significant increase in renin-like activity in the hypothalamus and fronto-parietal cortex in rats subjected to immobilization stress.84,91 Haulică and co-workers demonstrated that administration of saralasin (AT2 receptor antagonist) blocks stress-induced analgesia (assessed in terms of increase in latencies to thermoalgesic stimuli) in rats subjected to immobilization stress, suggesting the involvement of cerebral RAS in stress analgesia. 84 Raghavendra and co-workers reported the attenuation of immobilization stress (1 h)-induced analgesia (in terms of tail flick latency) with losartan (10 and 20 mg/kg i.p.), again suggesting the key role of RAS in producing analgesia. 86
3.2 Clinical studies
Clinical studies documenting the hyperalgesic actions of the RAS inhibitors have suggested the analgesic role of AngII. 29 It has been very well reported that hypertensive patients have reduced pain sensitivity, which is antagonized by the different RAS inhibitors, suggesting the anti-nociceptive actions of endogenous angiotensin peptides.92–94 Guasti and co-workers demonstrated that 8-week treatment with enalapril and losartan increases the pain sensitivity (in terms of dental pain threshold and tolerance) in 22 hypertensive patients with dental pulp stimulation. Furthermore, reduced perception of painful stimuli was reported in untreated hypertensives as compared with normotensives, indicating the anti-nociceptive effects due to activation of RAS during hypertension. 95 Another clinical study also evaluated the effects of ramipril and losartan on pain perception in 30 healthy normotensives using cola caps and handcuff of sphygmomanometer. No significant changes in pain threshold were reported in the control group, but the group receiving either ramipril or losartan showed a decline in threshold for maximum tolerated pain. The study also revealed that a single dose treatment of healthy volunteers with ramipril and losartan produces algesia as early as after ingestion of the first dose, suggesting the hypoalgesic and pain-attenuating effects of the RAS. 96 A clinical study has shown the strong association between the angiotensin neuropeptides and onset of complex regional pain syndrome (CRPS). Long-term treatment with ACE inhibitor was strongly associated with CRPS onset, which was mainly attributed to ACE inhibitors-mediated modulation of neuro-inflammatory mechanisms (alteration of substance P and bradykinin catabolism) involved in the pathophysiology of CRPS.29,97 The development of intestinal angioedema caused by ACE inhibitors such as lisinopril is also very well documented in the literature.98–101
3.3 Possible mechanisms
3.3.1 Opioids
The analgesic actions of AngII may be possibly mediated directly through endogenous opioids in the body. Studies based on stress-induced analgesia have demonstrated that AngII- and stress-induced analgesic actions are abolished in the presence of naloxone (an opioid antagonist), suggesting that the cerebral RAS participates in stress analgesia indirectly by releasing endogenous opioid peptides.84–86 Studies in the SHR have also suggested the role of endogenous opioids in producing the analgesic actions of RAS, as administration of naloxone was shown to attenuate an increased pain latency behavior in the SHR. 102 Pharmacological research has demonstrated that the RAS components are localized on the opioid-rich brain areas, including the PAG and trigeminal nucleus of the spinal cord.103–105 Furthermore, AngII has been shown to induce the release of β-endorphins in the brain.106–108 Therefore, it may be possible that peripheral AngII may increase the release of endogenous opioids from the sympathetic ganglia or facilitate the effectiveness of the endogenous opioid system to produce analgesic actions. 90
3.3.2 AT2 receptors
The anti-nociceptive actions of AngII may also be ascribed secondary to activation of the brain localized AT2 receptors, particularly in the PAG region, 16 as administration of the AT2 receptor antagonist PD123319 is shown to abolish ICV AngII-induced anti-nociceptive actions. 15 Another study has shown that the anti-nociceptive effects of AngII micro-injection in the ventrolateral PAG are abolished in the presence of AT2 receptor antagonist. 30 An acute administration of CGP42112A (AT2 receptor agonist) into the lateral ventricle has been shown to induce anti-nociceptive effects (assessed by the paw pressure test) in rats, suggesting that the activation of AT2 receptors is associated with analgesic actions. 102 On the contrary, in the same study, the chronic activation of AT2 receptors with CGP42112A was shown to produce nociceptive effects. The authors attempted to define CGP42112A-induced increased pain sensitivity on lines with morphine (gold standard for the treatment of acute pain)-induced hyperalgesia on its prolonged administration, and the changes in the receptor sensitivity (desensitization of AT2 receptors) was described as the possible mechanism responsible for an increased pain sensitivity with prolonged activation of AT2 receptors.102,109 CGP42112A peptide is a highly selective AT2 receptor ligand, which displaces AngII analogs by this receptor type in different brain structures.110,111 Most of the studies have described CGP42112A as a selective peptide AT2 receptor agonist.112–114 However, a few studies have also considered it as an AT2 receptor antagonist and employed it to abolish the effects of angiotensin neuropeptides. 115 It has been described as acting as a selective AT2 receptor ligand (may act as agonist as well as antagonist) depending upon the dose. 116
Furthermore, per se administration of PD123319 (single ICV dose of 5 µg/rat and multiple subcutaneous doses of 10 mg/kg/day/14 days) in normal animals has been shown to aggravate nociception, suggesting that the endogenous brain localized basal AT2 receptors mediate the analgesic actions.15,102 The pain-attenuating role of AT2 receptors has been supported by a study showing an increased pain sensitivity and decreased brain β-endorphin levels in AT2 knockout mice as compared with the corresponding wild-type mice. 117 It probably suggests that AngII may activate the brain localized AT2 receptors to release endogenous opioids to exert analgesic actions. On the contrary, Smith and co-workers demonstrated the analgesic effects of single intravenous (1–10 mg/kg) or oral (5–10 mg/kg) bolus doses of the AT2 receptor antagonist S-enantiomer of EMA400 in the CCI model of neuropathic pain in rats, suggesting that the activation of AT2 receptor produces nociceptive effects. Furthermore, treatment with EMA401 is shown to inhibit the functional response of capsaicin in cultured human and rat dorsal root ganglion (DRG) neuron responses. 118 Presently, it is difficult to explain the differential role of AT2 receptors in pain development or attenuation. The majority of studies suggesting the analgesic actions of AT2 receptors have mainly described the role of the brain localized AT2 receptors.84,117 On the contrary, the analgesic actions of EMA400 mainly involve the antagonism of peripheral AT2 receptors because it does not appear to cross the blood–brain barrier. 119
3.3.3 Bradykinin
The development of pain symptoms with ACE inhibitors may be linked to inhibition of the dipeptidylcarboxypeptidase enzyme, which is involved in the conversion of angiotensin I to II and degradation of kinins like bradykinin and substance P (potent mediators of inflammation and pain). Accordingly, ACE inhibitors may facilitate the actions of bradykinin (algesic peptide) due to its decreased metabolism, leading to development of pain. 120 The nociceptive role of bradykinin, one of the most potent endogenous neuropeptides, and its G-protein-coupled B1 and B2 receptors is very well documented. 121 Systemic administration of B1 receptor antagonist (Lys-[Des-Arg9,Leu8]-bradykinin) has been shown to reduce thermal and mechanical hypernociception in the sciatic nerve constriction and spinal cord injury models. 122 The knockout of B1 receptor genes is associated with attenuated pain perception in the sciatic nerve injury in mice. 123 Furthermore, administration of B1 receptor antagonists such as R-715, SSR240612, des-Arg(9)-bradykinin, LF22-0542 and B2 receptor antagonists such as HOE140, LF16-0687 attenuates pain in the brachial plexus avulsion and SNL models,124,125 suggesting a critical role for both B1 and B2 receptors in neuropathic pain. In addition to direct effects of bradykinin on pain, it also participates indirectly by stimulating the release of substance P from sensory nerves. 126 Furthermore, ACE also degrades substance P;127,128 therefore, the levels of substance P are increased directly due to ACE inhibition or indirectly due to bradykinin. The nociceptive role of substance P and neurokinin 1 receptor is very well documented. 129 Studies using knockout mice and selective NK1 receptor antagonists have revealed that the substance P–NK1 system contributes significantly in the sensitization of spinal neurons in neuropathic pain. In the CCI model, NK receptor antagonist (RP67580) has been shown to exhibit analgesic effects in a neuropathic pain model. 130
4. Possible hypothesis for dual actions of the RAS
As in pain studies, the dual role of AngII has also been described in stress-related anxiety9,10,131 and cognition.5,7,8 Studies based on pharmacological antagonism of RAS have mainly described the stress-inducing role of angiotensin.9,132 On the other hand, studies employing TGR(ASrAOGEN)680, within an active central angiotensin system, have shown the stress-attenuating role of endogenous angiotensin.133,134 Similarly, the actions of the angiotensin system on memory formation/enhancement as well as in memory impairment have also been described. 7 Our own studies have described the dual role of angiotensin neuropeptides in cognition.8,135 There have been contradictory results, i.e. both pain attenuation and increased pain sensitivity with RAS blockers.28–30 However, it is difficult to explain the exact mechanism, as there are no direct studies to understand the duality of angiotensin neuropeptides in pain. The evidence regarding the analgesic role of RAS blockers has mainly come from preclinical and clinical studies in which inflammation is a predominant feature (e.g. neuropathic pain and migraine).39,40 The ability of RAS blockers to attenuate angiotensin-mediated amplification of the inflammatory milieu has been considered as the major mechanism responsible for decreased pain perception. On the other hand, most of the clinical studies documenting the increased pain perception with RAS blockers are in either normal or hypertensive patients in whom inflammation reactions are not set up. 96 The ability of ACE inhibitors (not AT1 antagonists) to decrease the breakdown of bradykinin has also been considered as the principal mechanism responsible for increased pain perception. Furthermore, most of the preclinical studies documenting the anti-nociceptive actions of AngII are based on its central (in the brain) administration. Activation of AT2 receptors plays a critical role in producing anti-nociception.16,30 It has been proposed that central AngII-mediated increase in opioidergic activity, possibly through AT2 receptor activation, may be responsible for decreased perception.84,86,90,117 Studies based on stress-induced analgesia have also described the critical role of central AngII in producing analgesia. 84 Nevertheless, further experimental studies are needed to explain the dual effects of angiotensin peptides in pain.
5. Role of other angiotensin neuropeptides in pain
Angiotensin-(1-7) (Ang-(1-7)) has also been considered as an important biologically active component of the RAS in the central nervous system; therefore the role of Ang-(1-7) in pain cannot be ignored. A study by Costa and co-workers has demonstrated the existence of Ang-(1-7) receptor (Mas-R) in the DRG and anti-nociceptive effect of Ang-(1-7) (4 μg/paw) in the rat paw pressure test. The specific antagonist for the Mas receptor, A-779, was shown to inhibit Ang-(1-7)-induced anti-nociception in a dose-dependent manner. However, administration of naloxone was unable to inhibit the anti-nociceptive effects of Ang-(1-7), suggesting that it plays an important role in modulating pain perception via Mas receptor activation in an opioid-independent pathway. 136 Another study has shown the existence of Mas-R within the dorsolateral PAG (dl-PAG), and administration of 100 nM Ang-(1-7) was shown to reduce the discharge rates of dl-PAG neurons (indicative of pain attenuation) in a whole cell patch-clamp recording. 137 Pelegrini-da-Silva and co-workers have explored the anti-nociceptive action of AngIII in rat ventrolateral PAG (vlPAG) using peptidase inhibitors and receptor antagonists in the tail-flick and incision allodynia models. AngIII injection into the vlPAG has been shown to produce anti-nociceptive effects (with increase in tail-flick latency), which in turn were blocked by losartan and CGP42,112A, but not by divalinal-AngIV, indicating that the effects of AngIII are mediated byAT1 and AT2 receptors, but not by the AT4 receptor. Furthermore, amastatin (AM), an aminopeptidase A inhibitor, is shown to inhibit the formation of AngIII from AngII and block the anti-nociceptive actions of AngII injection into the vlPAG, suggesting that conversion of AngII to AngIII in the vlPAG is required to elicit anti-nociception and that it can be ascribed to AngIII. 115
Conclusion
Various preclinical and clinical studies have documented the dual role of the renin–angiotensin–aldosterone system in pain. The pain-attenuating action of renin–angiotensin–aldosterone blockers has been linked to decreased cytokine synthesis and inflammatory load. On the contrary, the pain-inducing actions of ACE inhibitors have been ascribed to interference with catabolism of bradykinin and substance P. Nevertheless, further studies are needed to unfold the complexities regarding the role of the renin–angiotensin–aldosterone system in the pathophysiology of pain.
Footnotes
Acknowledgements
The authors are grateful to the Department of Pharmaceutical Sciences and Drug Research, Punjabi University, Patiala, India for supporting this study.
Conflict of interest
The authors declare that they have no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.