
Abstract
Introduction:
Angiotensin-converting enzyme (ACE) inhibitors cause angioedema due to diminished degradation of bradykinin. Angiotensin receptor blockers may occasionally cause angioedema but the mechanism is unknown, and are generally considered safe, even in those who have reacted to ACE inhibitors. We determined whether aliskiren, a renin inhibitor, has an effect on the rate of bradykinin degradation.
Methods:
The ability of renin to metabolize bradykinin was studied and the rate of bradykinin degradation compared in the presence or absence of aliskiren. Enalapril, a known ACE inhibitor that causes angioedema served as positive control.
Results:
Renin was unable to digest bradykinin, indicating that a renin inhibitor is unlikely to affect the rate of bradykinin degradation. In a plasma system, aliskiren had no effect on the rate of bradykinin degradation while enalapril inhibited it appreciably. An inhibitory effect of aliskiren on the rate of bradykinin degradation by human pulmonary endothelial cells was observed, estimated to be about 5% of that of enalapril.
Conclusion:
Aliskiren has no effect upon the rate of bradykinin degradation in plasma and a minimal effect employing vascular endothelial cells. The latter suggests inhibition of a non-renin enzyme that is a minor contributor to bradykinin degradation.
Introduction
Angiotensin-converting enzyme (ACE) inhibitors cause angioedema by excessive accumulation of bradykinin due to an inhibited rate of degradation. The metabolism of brady-kinin occurs along endothelial cells within the pulmonary vasculature, as well as within blood plasma, and in each instance, ACE has a major role. 1 Bradykinin in plasma is degraded by carboxypeptidase N (kininase I) 2 which removes C-terminal arg. The product, termed des-arg9 bradykinin is inactive on constitutively present B-2 receptors (the primary receptor responsible for angioedema), yet it is not totally inactive, since des-arg9 bradykinin can stimulate B-1 receptors induced during inflammation by cytokines such as interleukin I. 3
ACE in plasma is known as kininase II and removes phe-arg 4 from the C-terminal end of bradykinin. The residual heptapeptide is inactive on both B-2 and B-1 receptors. A second cleavage by ACE removes ser-pro, leaving the pentapeptide, arg-pro-pro-gly-phe. If des-arg9 bradykinin has been produced first, ACE will cleave ser-pro-phe from the C-terminal end which eliminates interaction with the B-1 receptor. 5 The role of additional enzymes in plasma (aminopeptidase P, neutral endopeptidase, dipeptidyl peptidase IV) purported to contribute to bradykinin metabolism (and perhaps contribute to the occurrence of angioedema)6-8 is unclear because in plasma, all the bradykinin is degraded to the pentapeptide arg-pro-pro-gly-phe plus ser-pro, phe-arg or ser-pro-phe before any other bonds are cleaved. 9 Thus all the initial products are the result of the action of the kininases I and II, while final degradation products are dipeptides or individual amino acids.
Digestion of bradykinin along the vascular endothelial surface 10 has actually not been studied in humans, although in vivo animal studies suggest a major contribution by ACE. Carboxypeptidase M at the cell surface is functionally indistinguishable from plasma carboxypeptidase N. 11 Here too, the role (if any) of aminopeptidase P, neutral endopeptidase, and dipeptidyl peptidase IV is not clear, although their functions, when studied individually, are well known.
The renin inhibitor aliskiren is marketed for the treatment of hypertension and acts to prevent cleavage of angiotensinogen to angiotensin I. 12 With angiotensin I formation being limited, there is no substrate for ACE, and angiotensin II levels decline. This appears to explain its efficacy in the treatment of hypertension. Since ACE digests angiotensin I and bradykinin, ACE inhibition can lead to elevated bradykinin levels which can result in cough or angioedema in a susceptible population. Aliskiren has no known role in bradykinin metabolism. Yet the FDA instructions regarding its use have indicated severe angioedema as a possible side-effect by analogy to the incidence observed with ACE inhibitors. This manuscript describes the ability of human plasma and pulmonary vascular endothelial cells to metabolize bradykinin and compares inhibition of bradykinin degradation seen by ACE inhibitor (as a positive control) with aliskiren.
Methods
Degradation of bradykinin using purified proteins
Bradykinin (0.1 µmol/l; Sigma-Aldrich, St. Louis, MO, Catalog number B-3259, ≥98% purity) was incubated with ACE (2 nmol/l), renin (9 U/ml), ACE plus enalapril (0.2 mmol/l; enalapril maleate salt, Sigma-Aldrich, St. Louis, MO, Catalog Number E6888, ≥98% purity), ACE plus aliskiren (0.2 mmol/l; supplied by Novartis pharmaceuticals), or renin plus aliskiren in HEPES buffered saline (HBS) for 2 h at room temperature. Samples were withdrawn at 2 min, 5 min, 10 min, 30 min, 60 min, and 120 min and bradykinin levels were determined by enzyme immunoassay (EIA). The proteins in the samples were precipitated with ice-cold ethanol, centrifuged for 30 min at 10,000 rpm in a microcentrifuge at 4°C and the supernatant containing free bradykinin was then evaporated to dryness using a centrifugal concentrator, and resuspended in the EIA buffer. The bradykinin EIA was performed using an assay kit obtained from Peninsula laboratories (Cat. No. S-1135, San Carlos, CA). The color intensity is inversely proportional to the amount of bradykinin present. It should be noted that in addition to bradykinin, lys-bradykinin, [Des-Arg1]-bradykinin and bradykinin-(4-9) are identified by this ELISA. The concentration of the peptide present in samples was calculated using a standard curve.
Degradation of bradykinin in plasma
Normal plasma was collected using citrate as anticoagulant and incubated with a 0.1µmol/l of bradykinin in the presence or absence of various concentrations of aliskiren (0.2 mmol/l) or enalapril (0.2 mmol/l). It should be noted that the potency of enalapril in plasma is increased relative to that of the parent compound because deesterification converts it to enalaprilat whose IC50 for ACE is 1000-fold lower, that is, a change from 1200 nM to 1.2 nM.13,14 Samples were withdrawn and processed as given above.
Degradation of bradykinin on vascular endothelial cells
Normal human pulmonary arterial endothelial cells (PAEC, cat. number CC2530) were obtained from Lonza Corporation (Walkersville, MD) and cultured in EGM-2 Bullet kit containing 2% fetal bovine serum (growth medium). For bradykinin degradation experiments, cells were grown in 96-well plates with 200µl growth medium. When confluent, cells were changed to serum-free media overnight and washed with HBS before the experiments. Cells were identified as endothelial cells by morphology as well as positive testing for von Willebrand factor. All the studies were done using cells at the tenth passage.
Results
We first incubated bradykinin (0.1 µmol/l) with 2 nmol/l ACE and determined its rate of degradation. Digestion was complete by the 1 h point (Figure 1(a)). When the experiment was repeated with 0.2 mmol/l of enalapril, bradykinin degradation was completely inhibited. We next performed the same experiment using renin in place of ACE. No degradation of bradykinin was observed (Figure 1(b)) and addition of the renin inhibitor aliskiren to the mixture had no effect. When we attempted to reverse ACE-catalyzed bradykinin degradation by addition of aliskiren in place of enalapril, no effect was observed (Figure 1(c)). These results demonstrate that renin does not degrade bradykinin and the aliskiren has no ACE-inhibitory effect.

Inhibition of bradykinin degradation in a purified system. Bradykinin (0.1 µmol/l) was incubated in HEPES buffered saline (HBS) alone or with (A) ACE (2 nmol/l) or ACE plus enalapril (ENA, 0.2 mmol/L), (B) renin (9 U/ml, Sigma-Aldrich, St. Louis, MO, catalog number R2779, ≥98% purity) or renin plus aliskiren (0.2 mmol/l), and (C) ACE or ACE plus aliskiren for 2 h at room temperature. Samples were withdrawn at indicated time points and bradykinin extracted and assayed by enzyme immunoassay (ELISA). All experiments were done in duplicates and were repeated at least three times. The data presented are mean +/- standard deviation of at least 6 values.
The next series of experiments examined bradykinin degradation in normal human plasma and compared enalapril and aliskiren for inhibition of any of the kininases present. DL-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid (MGTA) was also studied; this is primarily a carboxypeptidase inhibitor (at low dose) but at higher concentrations can inhibit all kininases so that bradykinin degradation is nil. In Figure 2(a) we added bradykinin (0.1 µmol/l) to plasma and followed its degradation. By 10 min, it had reached undetectable levels. When MGTA was added at 1 mmol/l, an initial rise in bradykinin level beyond that which was added was seen. This is due to endogenous bradykinin formation, but is only observed when a significant kininase inhibitor has been added. After a peak was reached at 10 min, there was progressive bradykinin degradation but at a much slower rate than was seen in the absence of MGTA. Addition of aliskiren with or without MGTA had no effect on the rate of bradykinin degradation (Figure 2(b)). When we substituted enalapril for MGTA (Figure 2(c)) there was significant impairment of the rate of bradykinin degradation compared with no inhibitor. When MGTA was added to enalapril, bradykinin degradation was almost completely inhibited. Addition of aliskiren to enalapril, or to the mixture of enalapril and MGTA, had no discernible effect on the rate of bradykinin degradation. We then tested aliskiren at a 10-fold higher concentration, that is, 2 mmol/l and again found no effect on the rate of bradykinin degradation. These data are consistent with kininase I and II (carboxypeptidase N and ACE) as the major bradykinin-degrading enzymes in plasma and demonstrate that aliskiren does not affect bradykinin degradation when examined alone or mixed with other kininase inhibitors.

Inhibition of bradykinin degradation in plasma. Bradykinin (0.1 µmol/l) was incubated in normal human plasma alone or with (A) MGTA (1 mmol/l), (B) Aliskiren (0.2 mmol/l) or aliskiren plus MGTA, (C) enalapril (0.2 mmol/l) or enalapril plus MGTA, and (D) enalapril plus aliskiren or enalapril plus aliskiren plus MGTA for 2 h at room temperature. These experiments were done in duplicate and were repeated at least three times. The data presented are mean +/- standard deviation of at least 6 values.
The final set of experiments examined bradykinin degradation by a monolayer of pulmonary artery endothelial cells. In this series of experiments we determined the rate of degradation of bradykinin by endothelial cell-derived enzymes. We incubated cells with 0.1 mmol/l bradykinin to which was added either enalapril or aliskiren or a mixture of both tested at three different concentrations (0.2 mmol/l, 1 mmol/l, 2 mmol/l). At 0.2 mmol/l (Figure 3(a)) there was no effect upon the rate of bradykinin degradation by aliskiren, while enalapril significantly slowed the rate of degradation, and the curve obtained with a mixture of aliskiren and enalapril appeared similar to that of enalapril alone. At 1 mmol/l an inhibitory effect of aliskiren was observed although inhibition by enalapril was much greater. Addition of aliskiren to enalapril did not affect the rate of degradation beyond that of enalapril alone. It should be noted (although not shown) that aliskiren at 1 mmol/l had no effect on the degradation of bradykinin in plasma, that is, it is no different from what was seen at 0.2 mmol/l aliskiren. When the experiment employing endothelial cells was repeated with all putative kininase inhibitors at 2 mmol/l, aliskiren again had an inhibitory effect on the rate of bradykinin degradation, somewhat greater than at 1 mmol/l, and when added to enalapril, an acceleration of the rate of bradykinin degradation in comparison with the effect of enalapril alone was seen (Figure 3(c)).

Inhibition of bradykinin degradation on human pulmonary artery endothelial cells. Confluent monolayers grown in 96-well plates were changed to serum-free medium overnight, washed with HEPES buffered saline (HBS) and incubated in HBS with bradykinin (0.1 µmol/l) alone or with aliskiren (ALI), enalapril (ENA), or a combination of both at different concentrations (A, 0.2 mmol/l; B, 1 mmol/l; and C, 2 mmol/l) in a total volume of 100µl for 3 h at 37°C. These experiments were done in duplicate and were repeated at least three times. The data presented are mean +/- standard deviation of at least 6 values.
Because an effect of aliskiren was observed, we did a dose–response comparing the rate of bradykinin degradation at 1 h in the presence of aliskiren or enalapril or MGTA tested at a dose range from 0.1–4 mmol/l. As seen in Figure 4, at 1 mmol/l MGTA inhibited bradykinin degradation completely. At this concentration MGTA is a non-specific metalloprotease inhibitor and inhibits all kininases present including ACE. As the dose of enalapril is increased, there is increasing inhibition of bradykinin degradation which reaches about 90% at 2 mmol/l at the 1 h time point. There is also inhibition of bradykinin degradation with aliskiren which reaches 20% at 2 mmol/l and 30% at 4 mmol/l. The equivalent inhibition (20%) by enalapril was at 0.1 mmol/l, a difference of about 20-fold.
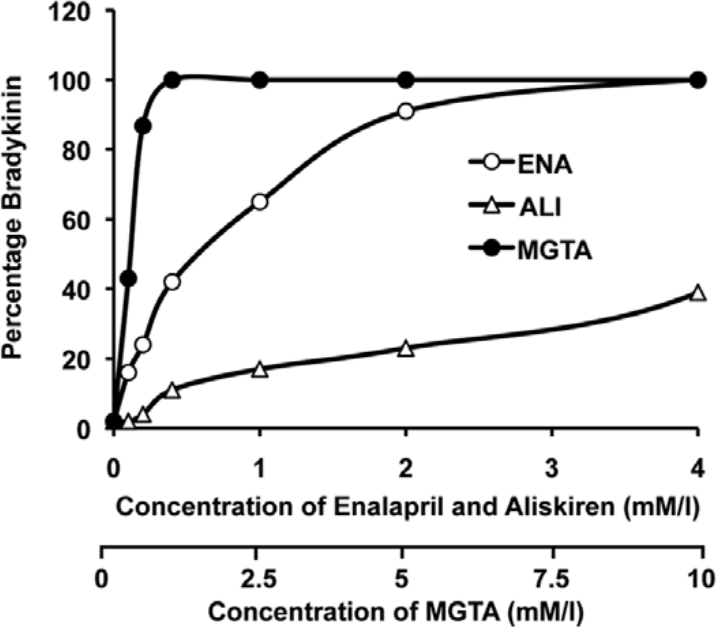
Concentration-dependent inhibition of bradykinin degradation by enalapril, aliskiren and mgta on human pulmonary artery endothelial cells. Confluent monolayers grown in 96-well plates were changed to serum-free medium overnight, washed with HEPES buffered saline (HBS) and incubated in HBS with bradykinin (0.1 mmol/l) in the presence of increasing concentration (0.1–4 mmol/l) of aliskiren (ALI), enalapril (ENA), or MGTA (0.1–10 mmol/l) in a total volume of 100 µl for 1 h and samples were collected and assayed for bradykinin by enzyme immunoassay (ELISA). The data presented are mean of two separate experiments.
Discussion
Angioedema due to ACE inhibitors is a well-described phenomenon which is caused by bradykinin accumulation due to inhibition of degradation by ACE. Although affecting only a small minority of those taking ACE inhibitors (incidence ~0.05%), the angioedema can be severe and life-threatening, particularly in African Americans where the incidence is five-fold greater than in Caucasians.15-17 This effect of ACE inhibitors is predictable because ACE is a key enzyme involved in bradykinin metabolism. Renin, by contrast, is not known to be a kininase and our data indicate no effect of renin on bradykinin metabolism. Thus a specific renin inhibitor should likewise have no effect. In fact, by inhibiting renin there is decreased angiotensin I formed, and ACE is the enzyme that converts angiotensin I to angiotensin II, which can raise blood pressure. As angiotensin I levels decrease, ACE will more readily degrade bradykinin since a competitive substrate has been depleted. The predicted effect of a renin inhibitor, therefore, is to augment the rate of bradykinin degradation rather than inhibit it. The incidence of angioedema in those taking aliskiren is no greater than a control population and is consistent with this prediction. 18
Nevertheless, while no effect of aliskiren is demonstrable in a plasma system at concentrations up to 2 mmol/l we find weak kininase inhibition (Figure 3(c)) when bradykinin is degraded by a pulmonary artery endothelial cell preparation with a potency we estimate to be about 5% of that of an ACE inhibitor. Since renin does not affect bradykinin degradation, we propose that aliskiren has an inhibitory effect on an endothelial cell-derived enzyme other than ACE or carboxypeptidase M. Possibilities include aminopeptidase P, neutral endopeptidase, and dipeptidyl peptidase IV, but action on an unknown kininase is more likely since all of these are present in plasma, where aliskiren has no effect. This resembles findings by Campbell et al. 19 in a rat system in which aliskiren was shown to have no effect on renin but did increase tissue kallikrein in the heart, an alternative source by which bradykinin could be produced. This is a possibility in our vascular endothelial cell experiments, although tissue kallikrein is not known to be produced by them. Liberation of heat shock protein 90 from such cells would activate any bound prekallikrein-HK complex 20 and produce plasma kallikrein. Experiments are in progress to further define this effect of aliskiren.
The inability of renin to degrade bradykinin, the absence of any kininase inhibitory activity of aliskiren in a plasma system, the very weak inhibitory activity observed employing vascular endothelial cells, and the absence of significant angioedema in a large study of patients using aliskiren indicate that aliskiren does not possess an effect resembling that of an ACE inhibitor. The incidence of angioedema in those taking the drug is not greater than the incidence in the population at large, and that remains true even if it is combined with an angiotensin receptor 1 inhibitor. 21 The effect seen with vascular endothelial cells is likely negated by the enhanced bradykinin degradation by ACE due to diminished levels of angiotensin I. There is no theoretical reason to believe that aliskiren would cause angioedema; clinical studies reveal no increased incidence of angioedema in those taking the drug, and the studies presented herein reveal no effect of aliskiren in a plasma system. Nevertheless, a study of aliskiren in the rat heart revealed an increase in bradykinin due to induction of tissue kallikrein. 19 Perhaps we are observing a similar effect employing vascular endothelial cells, but it does not appear to translate into any discernible clinical effect.18,21 When oral doses of the drug are taken by patients, the Cmax at 150 mg dose is 140 ng/ml, while at 300 mg it is 330 ng/ml. Our experiments were performed in the µg/ml range. Thus even the small effect observed employing vascular endothelial cells is of questionable physiologic significance given the absence of a significant increase in angioedema noted in patients utilizing the medication for treatment of hypertension. Thus physicians employing the drug for the treatment of hypertension should not anticipate an increase in the incidence of angioedema beyond that seen in the population at large, and the promulgation of warnings to that effect should be reconsidered.
Conclusions
Renin, in contrast to ACE, had no effect upon bradykinin metabolism when studied in vitro, that is, bradykinin was not degraded. Comparative studies of aliskiren, a renin inhibitor, vs. enalapril, an ACE inhibitor, on the rate of bradykinin degradation in human plasma revealed no effect of aliskiren at concentrations up to 1 mmol/l, while enalapril was readily shown to retard degradation. Bradykinin degradation by pulmonary vascular endothelial cells was prominently inhibited by enalapril at 0.2 mmol/l or 1 mmol/l, while aliskiren had no effect at 0.2 mmol/l and a small effect at 1 mmol/l. Further quantitations of the latter effect at concentrations up to 4 mmol/l revealed a 20-fold difference in inhibition of bradykinin degradation by enalapril compared with aliskiren, although the enzyme affected by aliskiren has not been identified.
Footnotes
Conflict of interest
None declared.
Funding
This study was funded by a grant (KJ) from Novartis Pharmaceuticals Corp., East Hanover, NJ.