
Abstract
Introduction:
We evaluate whether angiotensinogen AGT M235T (rs699), angiotensin-converting enzyme ACE (I/D) (rs4646994) and aldosterone synthase CYP11B2 –344C/T (rs1799998) polymorphisms can be genetic risk factors of chronic glomerulonephritis (GN) in the Polish population.
Materials and methods:
The study was conducted in 140 patients with primary chronic GN: mesangial proliferative GN (MesPGN) (n = 49), IgA nephropathy (IgAN) (n = 31), membranous nephropathy (MN) (n = 27), focal segmental glomerulosclerosis (FSGS) (n = 25), membranoproliferative GN (MPGN) (n = 4), and minimal change disease (MCD) (n = 4), and controls (n = 187). Genotypes were determined by HRM curve analysis for AGT M235T, by PCR and agarose gel separation for ACE (I/D), and by PCR-RFLP for CYP11B2 –344C/T.
Results:
We found a significant association of the CYP11B2 –344C/T polymorphism in the recessive model with all subtypes of GN (OR = 1.925 (95% CI = 1.152–3.219, p = 0.0118, pcorr = 0.0354)). We also observed that the CYP11B2 –344C/T polymorphism in the recessive model may also be an independent significant risk factor of IgAN (OR = 2.743 (95% CI = 1.219–6.172, p = 0.0122, pcorr = 0.0366)), FSGS (OR = 2.895 (95% CI = 1.200–6.985, p = 0.0145, pcorr = 0.0435)), and all proliferative GNs (MesPGN, IgAN, MPGN) (OR = 2.171 (95% CI = 1.211–3.894, p = 0.0084, pcorr = 0.0252)).
Conclusion:
Our results suggest that the CYP11B2 –344C/T polymorphism might be an independent risk factor of IgAN, FSGS and all proliferative chronic GNs.
Keywords
Introduction
Primary chronic glomerulonephritis (GN) is an inflammation of the filtering units of the kidney and frequently progresses to end-stage renal disease.1–3 The main renal lesions include glomerular sclerosis and tubulointerstitial injury, and result in outflow of blood cells and protein into the urine. 1 Kidney damage may be due to the patient’s immune response abnormalities, which can be triggered by infectious pathogens or drug and toxin exposure.1–3 The interaction between genetic background and environmental factors has been recognized in the development of GN.4–7 However, the precise etiologic agents in most glomerulonephritides (GNs) have not been identified.1–7
The action of the renin-angiotensin-aldosterone system (RAAS) in controlling blood pressure and homeostasis of sodium and fluids has been well described; 8 however, this system may also play a pathophysiologic role in the development and progression of GN. 8 The biologically active product of RAAS, angiotensin II (Ang II), elevates the glomerular hydraulic pressure, which may contribute to the development and progression of kidney diseases. 9 Binding of Ang II to its renal receptor, independent of the hemodynamic mechanism, may also directly elicit glomerular injury by promoting inflammatory processes, cell proliferation, and extracellular matrix synthesis. 9 Moreover, it has been suggested that aldosterone, which is similar to Ang II, may exert direct inflammatory and fibrotic actions, leading to podocyte damage and mesangial cell proliferation and promoting the onset and progression of GN. 10
Therefore, gene variants linked to changes in the expression of RAAS components and overactivity in this system may predispose to the onset of GN and to acceleration in the loss of glomerular filtration in patients with GN.11–14 Among the genes encoding the RAAS components, of particular interest are angiotensinogen (AGT), angiotensin-converting enzyme (ACE), and aldosterone synthase (CYP11B2). Single-nucleotide polymorphisms (SNPs) situated in these genes have been demonstrated to be risk factors for onset of and progressive renal dysfunction in various subtypes of GN.11–16 Studies have demonstrated that the 235T allele of the AGT M235T (rs699) polymorphism has been associated with higher plasma AGT levels. 17 An insertion/deletion of a 287-base pair in intron 16 of ACE (I/D) (rs4646994) has been shown to significantly affect circulating and tissue ACE levels and is responsible for an increased production of Ang II. 18 Moreover, the CYP11B2 –344C/T (rs1799998) polymorphism has been shown to influence aldosterone production.19,20 The AGT M235T, ACE (I/D), and CYP11B2 –344C/T polymorphisms have also been associated with an increased risk of renal diseases in some, but not all, studies.11–16,21,22
Therefore, we analyzed the distribution of polymorphic variants of AGT M235T, ACE (I/D), and CYP11B2 –344C/T in patients with various morphological types of primary chronic GN and controls in a sample of the Polish population.
Materials and methods
Patients and controls
One hundred and forty patients with primary chronic GN who underwent renal biopsy between October 2009 and Jun 2011 were randomly selected for the study at the Department of Nephrology, Transplantology and Internal Medicine, Poznań University of Medical Sciences. The patient group with primary chronic GN was composed of mesangial proliferative GN (MesPGN) (n = 49), IgA nephropathy (IgAN) (n = 31), membranous nephropathy (MN) (n = 27), focal segmental glomerulosclerosis (FSGS) (n = 25), membranoproliferative GN (MPGN) (n = 4), and minimal change disease (MCD) (n = 4) (Table 1). Controls were matched by age and gender to the patients and consisted of 187 unrelated healthy women and men sequentially selected from healthy volunteers (Table 1). Cases and controls were Caucasian and from the same geographic area of Poland. Written consent for study involvement was provided by all individuals. The procedures of the study were accepted by the local ethical committee of Poznań University of Medical Sciences.
Clinical characteristics of patients with primary chronic glomerulonephritis (GN).

mean ± standard deviation; bmedian (range).
Genotyping
DNA was isolated from peripheral white blood cells using a standard salting out procedure. Genotyping of ACE (I/D) (rs4646994) was conducted by polymerase chain reaction (PCR) followed by separation on 2% agarose gel (Supplemental data, Table 1, and Table 2). To avoid mistyping of the ACE (I/D) polymorphism, all samples with the DD genotype were subjected to PCR amplification with the use of insertion-specific primers (Supplemental data, Table 2). The presence of the AGT M235T (rs699) polymorphism was determined by high-resolution melting curve analysis (HRM) on the LightCycler 480 system (Roche Diagnostics, Mannheim, Germany) (Supplemental data, Table 1, and Table 2). Genotyping of the CYP11B2 –344C/T (rs1799998) polymorphic variant was assessed by PCR-restriction fragment length polymorphism (PCR-RFLP) conducted according to the manufacturer’s instructions (Fermentas, Vilnius, Lithuania) (Supplemental data, Table 1, and Table 2). DNA fragments were separated on 2% agarose gel and visualized by ethidium bromide staining. The genotyping quality of all polymorphisms was examined by repeat analysis of approximately 10% of the samples using the initial genotyping method or direct commercial sequencing.
Association of polymorphisms of ACE, AGT and CYP11B2 genes with primary chronic glomerulonephritis (GN).
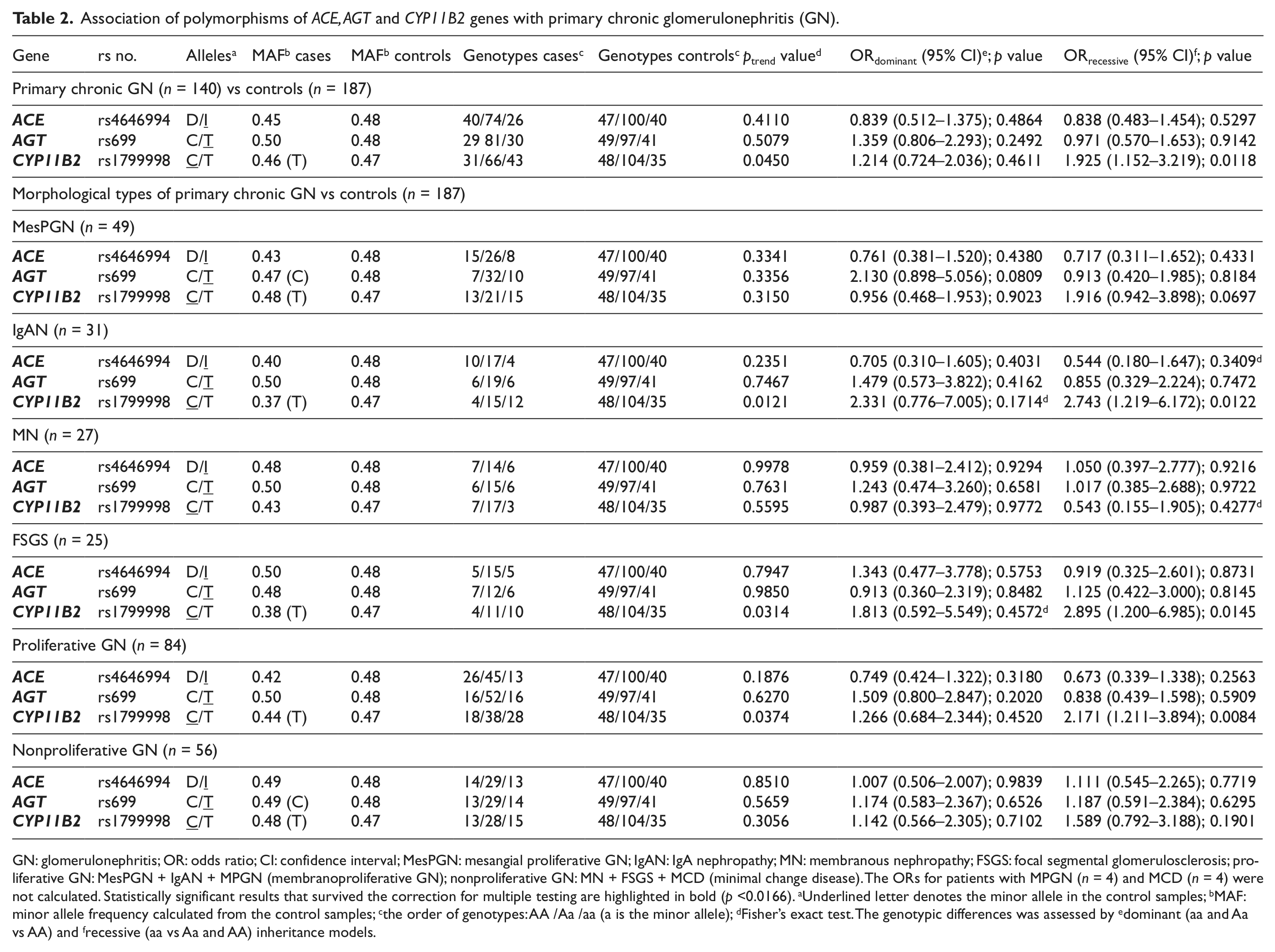
GN: glomerulonephritis; OR: odds ratio; CI: confidence interval; MesPGN: mesangial proliferative GN; IgAN: IgA nephropathy; MN: membranous nephropathy; FSGS: focal segmental glomerulosclerosis; proliferative GN: MesPGN + IgAN + MPGN (membranoproliferative GN); nonproliferative GN: MN + FSGS + MCD (minimal change disease). The ORs for patients with MPGN (n = 4) and MCD (n = 4) were not calculated. Statistically significant results that survived the correction for multiple testing are highlighted in bold (p <0.0166). aUnderlined letter denotes the minor allele in the control samples; bMAF: minor allele frequency calculated from the control samples; cthe order of genotypes: AA /Aa /aa (a is the minor allele); dFisher’s exact test. The genotypic differences was assessed by edominant (aa and Aa vs AA) and frecessive (aa vs Aa and AA) inheritance models.
Statistical analysis
The distribution of genotypes in primary chronic GN patients and controls was tested for deviation from the Hardy-Weinberg equilibrium. The chi-squared (χ2) or Fisher’s exact tests were used to evaluate differences in genotypic and allelic prevalence between patients with various subtypes of GN and controls. Moreover, the odds ratio (OR) and 95% confidence intervals (95% CI) were calculated. The data were analyzed under recessive and dominant inheritance models (aa vs Aa + AA and Aa + aa vs AA, respectively; where “a” is the minor allele). The polymorphisms were tested for association with primary chronic GN incidence using the chi-square test for trend (ptrend).
Results
Genotype analysis of the AGT M235T (rs699), ACE (I/D) (rs4646994), and CYP11B2 –344C/T (rs1799998) polymorphisms did not demonstrate a significant deviation from the Hardy-Weinberg equilibrium in patients and control groups. We found a significant association of the CYP11B2 –344C/T polymorphism in the recessive model with all primary subtypes of GN (OR = 1.925 (95% CI = 1.152–3.219, p = 0.0118, pcorr = 0.0354)), but this association was not present in the dominant model (OR = 1.214 (95% CI = 0.724–2.036, p = 0.4611)) (Table 2). We also observed that the CYP11B2 –344C/T polymorphism in the recessive model may also be an independent significant risk factor of IgAN (OR = 2.743 (95% CI = 1.219–6.172, p = 0.0122, pcorr = 0.0366)), FSGS (OR = 2.895 (95% CI = 1.200–6.985, p = 0.0145, pcorr = 0.0435)), and all proliferative GNs (MesPGN, IgAN, MPGN, Table 1) (OR = 2.171 (95% CI = 1.211–3.894, p = 0.0084, pcorr = 0.0252)) (Table 2). We also determined significance in the p values of the χ2 test for the trend observed for all primary subtypes of GN (ptrend = 0.0450), IgAN (ptrend = 0.0121), FSGS (ptrend = 0.0314), and all proliferative GNs (ptrend = 0.0374) (Table 2). However, we did not find a significant difference in the distribution of this polymorphism between controls and patients with MN, and MesPGN and all nonproliferative GNs (MN, FSGS, MCD, Table 1, and Table 2). Moreover, there was no significant difference in the prevalence of the AGT M235T and ACE (I/D) polymorphic variants between patients with all primary subtypes of GN or different morphological types of primary GN and controls (Table 2). We also did not observe an association between the studied polymorphisms with gender or clinical manifestations of GN.
Discussion
Aldosterone is one of the main effectors of the RAAS system, with its classical action being the regulation of Na+ and water retention and K+ excretion in the distal nephron and other epithelial tissues in normal physiology. 23 Aldosterone secretion is regulated primarily by plasma levels of Ang II and K+. 24 Aldosterone is produced in the adrenal zona glomerulosa cells, where Ang II and K+ induce expression of the CYP11B2-encoding terminal enzyme of aldosterone biosynthesis. 25 However, there has recently been increasing evidence of extra-adrenal biosynthesis of aldosterone. 26 Moreover, aldosterone may exert non-hemodynamic actions on nonepithelial cells in the cardiovascular system, where it may be responsible for hypertrophic and fibrotic effects, especially in patients with a high salt intake. 10 In the kidney, the non-hemodynamic actions of aldosterone may also promote pathological processes involved in the onset of renal disease in general, and especially in GN.10,27 Direct action of aldosterone in podocytes leads to their severe damage and denudation of the glomerular basement membrane. 28 In the kidney, aldosterone also increases the biosynthesis of intercellular adhesion molecule-1, interleukin-6, and monocyte chemoattractant protein-1, all of which are main mediators of inflammation and the progression of mesangial fibrosis.29,30 Moreover, aldosterone induction of biosynthesis of transforming growth factor-β promotes fibrotic effects in vitro in mesangial cells. 31 Activation of epidermal growth factor, and subsequent effects of mitogen-activated protein kinases, directed by aldosterone in vitro result in the proliferation of mesangial cells. 32 Moreover, studies conducted in animal models have demonstrated that aldosterone antagonists may reduce mesangial cell proliferation, proteinuria and tubule interstitial injury, inhibit podocyte damage, and lead to the regression of glomerulosclerosis.33,34,35 This beneficial effect of aldosterone antagonists has also been confirmed in patients with renal disease whose treatment reduced proteinuria and inhibited the progression of renal disease.36,37
Therefore, genetic variants of CYP11B2 that may be associated with increased biosynthesis of aldosterone may also be risk factors in the development of GN and in the progression of renal dysfunction in primary GN.
We found a significant association of the CYP11B2 –344 CC genotype with all studied types of primary chronic GN. Moreover, the CYP11B2 –344CC genotype was found to be an independent risk factor of some morphological types of primary chronic GN, such as IgAN, FSGS, and proliferative GN. However, the lack of association of CYP11B2 –344T/C with the other studied types of primary chronic GN might be due to the small sample size.
Recently, Bantis et al. (2011) demonstrated a significant association between the CYP11B2 –344C gene variant with the development of FSGS. 15 Moreover, Bantis et al. (2011) found a significantly increased CYP11B2 –344CC/CT genotype frequency in fast progressors over slow progressors in Caucasian patients with IgAN. 16 In addition to these findings, female patients with IgAN having the CYP11B2 –344 CC genotype displayed significantly poorer renal survival than those with other genotypes. 38 In contrast, Huang et al. (2010) demonstrated that CYP11B2 –344C/T polymorphism genotypes distribution was similar in patients with IgAN and controls from Chinese and Korean populations.11,21 There was also no association of the CYP11B2 –344C/T polymorphism with end-stage renal disease in patients with diabetic nephropathy. 39
We did not find an association between the AGT M235T and ACE (I/D) polymorphisms with either all or separate morphological types of the studied primary GNs. There are many studies that have demonstrated an association of the AGT M235T polymorphism with the progression of primary GN rather than with the development of GN.11,12,40,41,42 However, the ACE (I/D) polymorphism, in contrast to our findings, has been shown to be a risk factor of the onset of some different morphological types of primary GN.13,14 The differences in the effects of the studied polymorphisms on the susceptibility and progression of various morphological types of primary GN between disparate ethnicities may be due to exposure to distinct environmental factors, racial heterogeneity, and the size of the studied groups.7,8
The role of the CYP11B2 –344C/T polymorphism in the production of aldosterone in humans has been studied by several researchers.19,20,43–46 Pojoga et al. (1998), in a cohort of 216 hypertensive patients of European origin, demonstrated that bearers of the CYP11B2 –344C allele exhibited elevated levels of blood plasma aldosterone. 19 In another study of 562 Caucasian patients, blood plasma aldosterone levels were increased in the bearers of genotypes in the following decreasing order: TT, CT, and CC, but the differences in levels were not statistically significant. 43 However, other studies have demonstrated contrasting results indicating either higher blood plasma or urinary excretion of aldosterone in carriers of CYP11B2 –344T gene variants than in those not bearing this gene variant.20,44–46 There are no data indicating the effect of the CYP11B2 –344C/T polymorphism on aldosterone levels in tissues. Moreover, the CYP11B2 –344C gene variant has been associated with increased arterial blood pressure in some studies.44,47
The molecular mechanism of the −344T/C SNP responsible for the modulation of CYP11B2 transcription is obscure.25,48,49 Despite having a binding site for steroidogenic transcription factor 1 (SF-1), the CYP11B2 –344T/C gene variants do not directly affect transcription. 48 Clyne et al. (1997) suggested that binding of SF-1 to the −344T/C site makes the promoter less available for binding to other transcription factors required for both basal and Ang II- or K+-stimulated CYP11B2 transcription. 25 Recently, Iwai et al. (2007) used reporter analysis in vitro to demonstrate that only the CYP11B2 –344 C type promoter responds in the presence of Ang II. 49
In summary, aldosterone has been suggested as exerting a direct effect on the onset of GN. Our study demonstrates an association of the CYP11B2 –344T/C, but not AGT M235T or ACE (I/D) SNPs with some different morphological types of primary chronic GN. The genetic study confirms a previously demonstrated association of CYP11B2 –344T/C polymorphism with the onset of FSGS. 15 Because our study was conducted on a limited number of cases, it should be further repeated in a larger group, as well as in independent cases and control groups in other ethnicities.
Footnotes
Acknowledgements
The technical assistance of Sylwia Matuszewska is gratefully acknowledged.
Conflict of interest
The authors declare that there are no conflicts of interest.
Funding
This work was supported by grant number 502-01-01124182-07474, Poznan University of Medical Sciences.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.