
Abstract
Introduction:
Angiotensin II is an effector peptide showing multiple physiological effects, such as regulation of vascular tone, tissue growth and remodelling. Postlactational involution of mammary gland involves changes such as high matrix metalloproteinase activity and release of bioactive fragments of fibronectin and laminin, which may be directly regulated by angiotensin II. The aim of the present study was to evaluate the influence of angiotensin II on proliferation, viability and motility of normal human mammary epithelial cells (184A1 cell line) and to determine the role of angiotensin II receptors in these processes.
Materials and methods:
Real-time reverse transcription-PCR, western blot and gelatin zymography were used to study the effect of angiotensin II on the expression of angiotensin receptors and matrix metalloproteinases in 184A1 cells. WST-1, AlamarBlue and BrdU assays were used as indicators of cell viability and proliferation after angiotensin II stimulation. Boyden chamber assays and monolayer wound migration assay were used to evaluate in vitro the changes in cell adhesion, migration and invasion.
Results:
Angiotensin II increased motility of the 184A1 cells and the ability of wound closure. Modifications in cell–substrate adhesion systems and increased secretion and activity of matrix metalloproteinases were also observed. The effect of angiotensin II was abolished by blocking angiotensin type 1 receptor with specific inhibitors candesartan and losartan.
Conclusions:
The results indicate that angiotensin II modulates cell behaviour via AT1-R and stimulates secretion of MMP-2 by human mammary epithelial cells.
Introduction
Cell motility has been described as a main part of tissue development and remodelling. This phenomenon takes place during several physiological processes, such as embryonic morphogenesis and adult wound healing. This process is also an important component of some pathological conditions, such as invasion and metastasis of tumour cells. 1 Mammary gland microenvironment during postlactational involution shares similarities with inflammation, including high matrix metalloproteinase (MMP) activity, fibrillar collagen deposition and release of bioactive fragments of fibronectin and laminin. 2 The metastatic process observed in the case of breast cancer also involves cell adhesion, migration and invasion, which are mediated by cell surface receptors. 3 Molecules that mediate these processes include transmembrane glycoproteins that determine both cell–cell binding and the interaction between cells and extracellular matrix (ECM) proteins. 3
Angiotensin II (AngII) is the main effector peptide in the renin–angiotensin system (RAS). AngII has multiple physiologic effects that regulate vascular tone, hormone secretion, tissue growth and neural activity.4,5 It has systemic (endocrine) and local (paracrine and autocrine) effects, favouring cell growth and differentiation. It acts through four types of receptors, of which types 1 and 2 (AT1-R and AT2-R) are the most important. 6 Stimulation of AT1-R leads to activation of intracellular pathways, which finally leads to vasoconstriction, inflammation and proliferation. Several reports indicate that AngII can induce the expression of different growth factors. Among other mechanisms associated with AngII functions are regulation of metalloproteinase expression and induction of inflammation. Angiogenesis is a fundamental process in tissue repair and development, and it participates in several pathological processes. In addition, the AT1-R is expressed in many malignant neoplasms and its blockade through AngII antagonists has been shown to have an antineoplastic effect. 7 Additionally, the use of AngII antagonists causes inhibition of angiogenesis, which has been demonstrated on tumoral experimental models. 6 Interestingly, most of the major functions of AngII, such as angiogenesis, migration and inflammation, are also related to cancer progression. 7
Most components of the RAS are expressed locally in a large variety of tissues and tumours, including mammary gland and breast cancers.3,7-9 AngII is known to stimulate proliferation of breast cancer cells, but it may also have other activities. One possibility is that it may be involved in breast cancer progression. 3 Since local synthesis of AngII may be important for mammary tissue organization and remodelling, one can speculate that tissue RASs may be involved in these processes. It has been well established that action of AngII regulates expression of ECM proteins in various tissues. 10 It has been recently demonstrated that AngII induced AT1-R mediated breakdown of tissue basement membrane by increasing MMP-2 and MMP-9 activity in various premalignancies and tumours.7,10-12
So far, there have been no published studies investigating the role of AngII (and AT1-R antagonists candesartan and losartan) on migration and motility potential of non-cancer cells. Therefore, the present work was undertaken to investigate the possible pro-motility activity of AngII and the effect of blocking the AngII type 1 receptor using AT1-R antagonists, candesartan and losartan. The aim of this study was to evaluate the effects of AngII on cell migration, proliferation and viability of normal human mammary epithelial cells, as well as to determine the effect of AngII on angiotensin receptors.
Material and methods
Cell culture
Human mammary epithelial 184A1cell line was purchased from the American Type Culture Collection (LGC Standards Sp. z o.o., Poland). The 184A1 cells at passages 3 to 15 were plated at subconfluent density on 75 cm2 flasks and grown to confluence in maintenance medium according to recommendations comprising DMEM/Ham’s F-12 (1:1) medium supplemented with 5% (v/v) horse serum (Life Technologies Corporation) as previously described by Zhao and Kulkarni and colleagues.13,14 For the purpose of experiments, the maintenance medium was replaced with an assay medium (maintenance medium without phenol red) for 24 h. This medium was then replaced with starvation assay medium supplemented with 1% horse serum (HS) (Life Technologies Corporation) and experiments were performed after 24 h.
Cells were then treated with one of the following protocols:
Dose/time stimulation of cells with various AngII (Bachem, Poland) concentrations (0.1 nM, 1 nM, 10 nM, 100 nM and 1000 nM) for 1, 2, 6, 12 or 24 h;
Treatment of cells with 1000 nM AngII alone or in combination with 1 µM candesartan (Astra Zeneca), an AT1-R antagonist, 1 µM losartan (Adamed, Poland), an AT1-R antagonist, and/or a combination of both inhibitors for 1 h before AngII exposure for 12 or 24 h.
Cell viability and proliferation
Cell viability assays
Cell viability was determined by a WST-1 reagent (Roche Applied Science, Poland) and AlamarBlue (Life Technologies Corporation) according to the manufacturer’s instructions. To detect the effect of AngII and/or ATR inhibitors on cell viability, cells were seeded on 96-well plates at a density of 3 × 104 cells/well. After 24 h culture the cells were then treated as described above. At the end of the exposure period, 10 µl of the WST-1 or AlamarBlue (1:10 dilution) was added into each well and incubated for 2 h. Absorbance was measured on an ELX 808IU plate reader (BioTeck) at 450 nm with reference at 655 nm for WST-1 and 570 nm with reference at 600 nm for AlamarBlue. The analysis was performed in three independent experiments. The effect was expressed as: (optical density (OD) of treated cells/OD of non-treated cells) × 100.
Cell proliferation assay
Proliferation was measured by colorimetric immunoassay based on bromodeoxyuridine (BrdU) incorporation into the cellular DNA, following the instructions recommended by the manufacturer (Roche Applied Science, Poland). 15 The experimental design was analogous to those set for the WST-1 and AlamarBlue assays. The cells were incubated with BrdU labelling reagent for 4h, followed by fixation in a FixDenat solution for 30 min at room temperature. They were then incubated with a 1:100 dilution of anti-BrdU-POD for 2h at room temperature. Finally, the immune reaction was detected by adding the substrate solution, and the developed colour was measured at 450 and 650 nm in an absorbance microplate reader (ELX 808IU, BioTeck). The analysis was performed in three independent experiments. The effect of the experimental factors on cell proliferation was calculated as a percentage of cell proliferation measured in the non-treated cells.
Cell adhesion assay
Cells (3 × 103 cells in 1000 µl medium) were plated in sextuplicate on 24-well collagen type IV, laminin and fibronectin-coated (Becton Dickinson Labware, Immunogen, Poland) plates (the attachment plate) or clear-bottomed uncoated plates (positive control). The plates were incubated at 37°C in a humidified 95% air 5% CO2 environment for 1 h. After incubation, medium was removed and wells were gently washed with sterile PBS to remove unattached cells. The remaining cells adhering to the matrix proteins were fixed with 4% paraformaldehyde (15 min) and stained with 0.1% crystal violet. After 10 min, crystal violet was removed and wells were gently washed with sterile PBS. At the end, 200 µl of 10% acetic acid was added for extraction, and after 15 min the dye/solution mixture was transferred to a 96-well plate and absorption was measured at 560–590nm (ELX 808IU, BioTeck). The total experiment was performed in triplicate.
Cell migration assay
For the migration assay, 184A1 cells were seeded at a density of 5 × 104 cells per well onto 8 µm pore transwell 24-well inserts (Becton Dickinson Labware, Immunogen, Poland) (modified Boyden chamber assay). The lower chamber was filled with 500μl 1% HS cell culture medium containing the test factors. The cells were incubated for 12 h at standard conditions. After this time non-migrating cells that remained on the upper surface of the filter were removed with cottonwool; the cells that appeared on the lower surface of the filter were fixed in 4% paraformaldehyde stained with 0.1% crystal violet. To assess the stained cells, they were dissolved in 200 µl of 10% acetic acid and the dye/solute mixture was transferred to a 96-well plate and measured at 560–590 nm (ELX 808IU, BioTeck). The experiment was performed in triplicate.
In vitro invasion assay
184A1 cells were added to inserts (8 µm pore) coated with GeltrexTM (Life Technologies Corporation) according to the manufacturer’s instructions. Twenty-four-well individual inserts were coated with 100 µl with GeltrexTM in a final concentration of 200 µg/ml (modified Boyden chamber assay). Cells (1×104 per 300 ml medium) were added to the inserts, which were placed into companion plates filled with 500 µl cell medium. The plates were incubated at standard conditions (37°C, 5% CO2) for 24 h. Then, the non-invasive cells that remained on the upper surface of the filter were removed with cottonwool; the cells that appeared on the lower surface of the filter were fixed in 4% paraformaldehyde stained with 0.1% crystal violet. To assess the stained cells, they were dissolved in 200 µl of 10% acetic acid, and the dye/solute mixture was transferred to a 96-well plate and measured at 560–590nm. The experiment was performed in triplicate.
Monolayer wound migration assay
184A1 cell were seeded onto a six-well plate and left to grow to 80% confluence. Cell monolayers were ‘wounded’ by scraping with a sterile 100µl pipette tip, forming approximately 0.5 mm gaps in the monolayers. Each well was then washed three times with PBS to remove the floating cells. Cells were stimulated by AngII (1000 nM) and/or inhibitors (1 µM). To differentiate the contribution of cell proliferation and migration to wound closure, cell cycle blocker hydroxyurea (0.5 mM) was added in the scratch wound model, according to a previously described method. 16 Images were collected by encoding the x, y coordinates for each wound location, which enabled the stage to return to the exact location of the original wound throughout the migration experiments. 17 Wound area measurements were averaged from three fields of the same well, using the ImageJ1.34s program (Wayne Rasband, National Institutes of Health, USA; http://rsb.info.nih.gov/ij/), and mean values obtained were taken as single data points. The effect of various agents on the areas covered by cells was calculated and is presented as a percentage of changes compared with control. 17
Gelatin zymography
Metalloproteinase activity was determined as previously described by Pons et al. 10 184A1 human cells (5 × 104) were cultured in six-well plates. The cells at 80% confluence were rinsed twice with Ca2+- and Mg2+-free PBS and incubated in conditional medium with AngII and/or ATR inhibitors. After 24 h incubation, conditioned culture medium was collected and clarified by centrifugation at 15,000 × g for 30 min at 4°C and stored at −80°C. Protein concentration was determined using Qubit® Protein Assay Kit (Life Technologies Corporation) and MMP-2/MMP-9 activity assessed using 10% gelatin zymography gels. Protein extracts (10 µg) were subjected to electrophoresis (120 V for 2 h) and subsequently incubated in 2.5% Triton X-100 for 1.5 h. Gels were next incubated in 50 mmol/l Tris buffer overnight, enabling determination of total proteolytic MMP activity with no interference from their associated tissue inhibitors. Gels were stained with Coomassie blue (Sigma-Aldrich, Poland) and destained with 50% methanol and 20% acetic acid. Areas of enzymatic activity appeared as clear bands over the dark background. 18 Gels were scanned using HP Precision ScanLT software, version 1.2, in a grey scale mode at 600 mm pixel size and 1708–1151 (X–Y) pixel count, using the autodensity feature on a scale ranging from 0 (clear) to 255 (opaque). The image was digitally inverted so that the integration of bands was reported as positive values. The pixel density was determined after background subtraction and used to calculate the integrated density of a selected band. Values of integrated density were reported in volume units of pixel intensity per mm2. Densitometry was performed using ImageJ 1.34s software (Wayne Rasband, National Institutes of Health, USA; http://rsb.info.nih.gov/ij/). The integrated density of each band is reported as the mean of three different measurements of the same gel for each sample run in triplicate. 19
Gene expression analysis
Total RNA was obtained using the TRIzol reagent, as described previously. 8 Isolated RNA samples were dissolved in RNase-free water, and RNA quantity was measured with the use of Qubit® RNA Assay Kit (Life Technologies Corporation). cDNA was synthesized from 10 µg of total RNA at a volume of 100 µl using ImProm RT-IITM reverse transcriptase (Promega, Poland) according to the manufacturer’s instructions. cDNA samples were diluted with sterile deionized water to a total volume of 100 μl and 2 μl was added to a PCR reaction. Real-time reverse transcription-PCR (RT-PCR) was performed using a Light Cycler 480 II (Roche, Poland). We analysed the relative expression of genes: AT1-R, AT2-R, MMP-2 and MMP-9. Their expression level was normalized to the mean expression of four reference genes (BMG2, RPS17, RPLPO, H3F3A).20,21 The primers were designed using Primer3 software (http://frodo.wi.mit.edu/). The following sequences of oligonucleotides were used in the experiments:
AT1-R F: 5′ ATT CGA CCC AGG TGA TCA AA 3′ AT1-R R: 5′ CCA CCA AGC TGT TTC CAA AT 3′; AT2-R F: 5′ GCA GCC TGA ATT TTG AAG GA 3′ and AT2-R R: 5′ ACC GCT GGT AAT GTT TTT GC 3′; MMP-9 F: 5′AAG TAC TGG CGA TTC TCT GAG GG; MMP-9 R: 5′GGC TTT CTC TCG GTA CTG GAA GAC; MMP-2 F: 5′TTT TCT CGA ATC CAT GAT GG; MMP-2 R: 5′CTG GTG CAG CTC TCA TAT TT; H3F3A F: 5′-AGG ACT TTA AAA GAT CTG CGC TTC CAG AG-3′; H3F3A R: 5′-ACC AGA TAG GCC TCA CTT GCC TCC TGC-3′; RPLPO F: 5′-ACG GAT TAC ACC TTC CCA CTT GCT AAA AGG TC-3′; RPLPO R: 5′-AGC CAC AAA GGC AGA TGG ATC AGC CAA G-3′; RPS17 F: 5′-AAG CGC GTG TGC GAG GAG ATC G-3′; RPS17 R: 5′-TCG CTT CAT CAG ATG CGT GAC ATA ACC TG-3′
All analyses were performed using a LightCycler FastStartDNA Master SYBR Green I kit (Roche Diagnostics) according to the procedure provided by the producer.
Western blot analysis
Total protein extracts were isolated using the RIPA protein extraction buffer consisting of 50mM Tris-HCl, 150mM NaCl, 0.5% NaDoc, 0.1% NP-40, 0.1% SDS and 2mM EDTA, supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich, Poland). The lysates were centrifuged at 14,000 × g and 4°C for 20 min and the pellets were discarded. Lysates from 184A1 cells were harvested and protein concentration was determined by the Bradford method (Bio-Rad, Poland) as described previously.4,22,23 Protein extracts (30 µg) were denatured in Laemmli’s sample buffer (bromophenol blue, 40% glycerol, 8–10% SDS, 20% β-mercaptoethanol), followed by 3 min heat denaturation at 100°C.The samples were then resolved by SDS-PAGE on a 12.5% polyacrylamide gel. After electrophoresis (120 V for 2 h), proteins were transferred in transfer buffer to a PVDF membrane using constant current (400 mA for 70 min). Transfer buffer was prepared by mixing 100 ml of 10× transfer buffer (200 mM Tris base, 1.5M glycine), 200 ml of methanol and 700 ml of water. Membranes were blocked in 5% non-fat dry milk–TBST solution (20 mM Tris-HCL, 500 mM NaCl, 0.5% Tween20) for 1 h at room temperature. Blots were incubated overnight at 4°C in blocking buffer containing one of the following primary antibodies: anti-AT1-R (sc-1173), anti-AT2-R (sc-9040) and anti-GAPDH (sc-59540) (Santa Cruz Biotechnology, AMX, Poland). Membranes were washed three times (3 × 15 min) with TBST, then incubated with Alkaline Phosphatase linked donkey anti-mouse or anti-rabbit antibody (Sigma-Aldrich, Poland) for 1 h at room temperature, and then washed three times (3 × 15 min) in TBST. Bands were visualized on the membranes, detection was performed using Novex® AP Chromogenic Substrate (BCIP/ NBT). Membranes were scanned using HP Precision Scan LT, in a grey scale mode at 600 mm pixel size and 1708–1151 (X–Y) pixel count, using the autodensity feature on a scale ranging from 0 (clear) to 255 (opaque). The pixel density was determined after background subtraction and used to calculate the integrated density of a selected band. Values of integrated density were reported in volume units of pixel intensity per mm2. A densitometric analysis of protein levels was performed with ImageJ 1.34s software (Wayne Rasband, National Institutes of Health, USA; http://rsb.info.nih.gov/ij/). The results were normalized to a reference protein: glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Statistical analysis
Statistical analyses were conducted using GrafPad Prism Software. Data in bar graphs (mean ± SEM) were analysed using Student’s t-test. p<0.05 was considered statistically significant.
The PCR array data were analysed by the ΔΔCt method. 24 An average of housekeeping genes BMG2, RPS17, RPLPO, H3F3A was used to obtain the ΔCt value for each gene of interest. The ΔΔCt value for each gene was calculated as the difference between the ΔCt of the treated groups and that of the control. The fold-change for each gene was calculated by the 2-ΔΔCt method.
Results
Cell viability and proliferation
In order to determine the time/dose response effect of AngII on cell viability and proliferation 184A1 cells were treated with AngII (0.1 nM, 1 nM, 10 nM, 100 nM and 1000 nM) for different time periods and the tested parameters were assessed by the WST-1, AlamarBlue and BrdU assays. AngII increased cell viability (WST-1, AlamarBlue assays) in a dose- and time-dependent manner. A statistically significant difference between the control and treated cells was observed only at two time points: 12 and 24 h.
AngII had a significant effect on viability of 184A1 cells with a maximal response obtained with 100 nM AngII (74.2 ± 4.9%; p<0.05) and 1000 nM AngII (70.1 ± 8.3%; p<0.05) at 12 h (Figure 1(a)) and at 24 h with 100 nM AngII (66.9 ± 6.1%; p<0.05) and 1000 nM AngII (72.9% ± 4.7%; p<0.05) (Figure 1(c)).
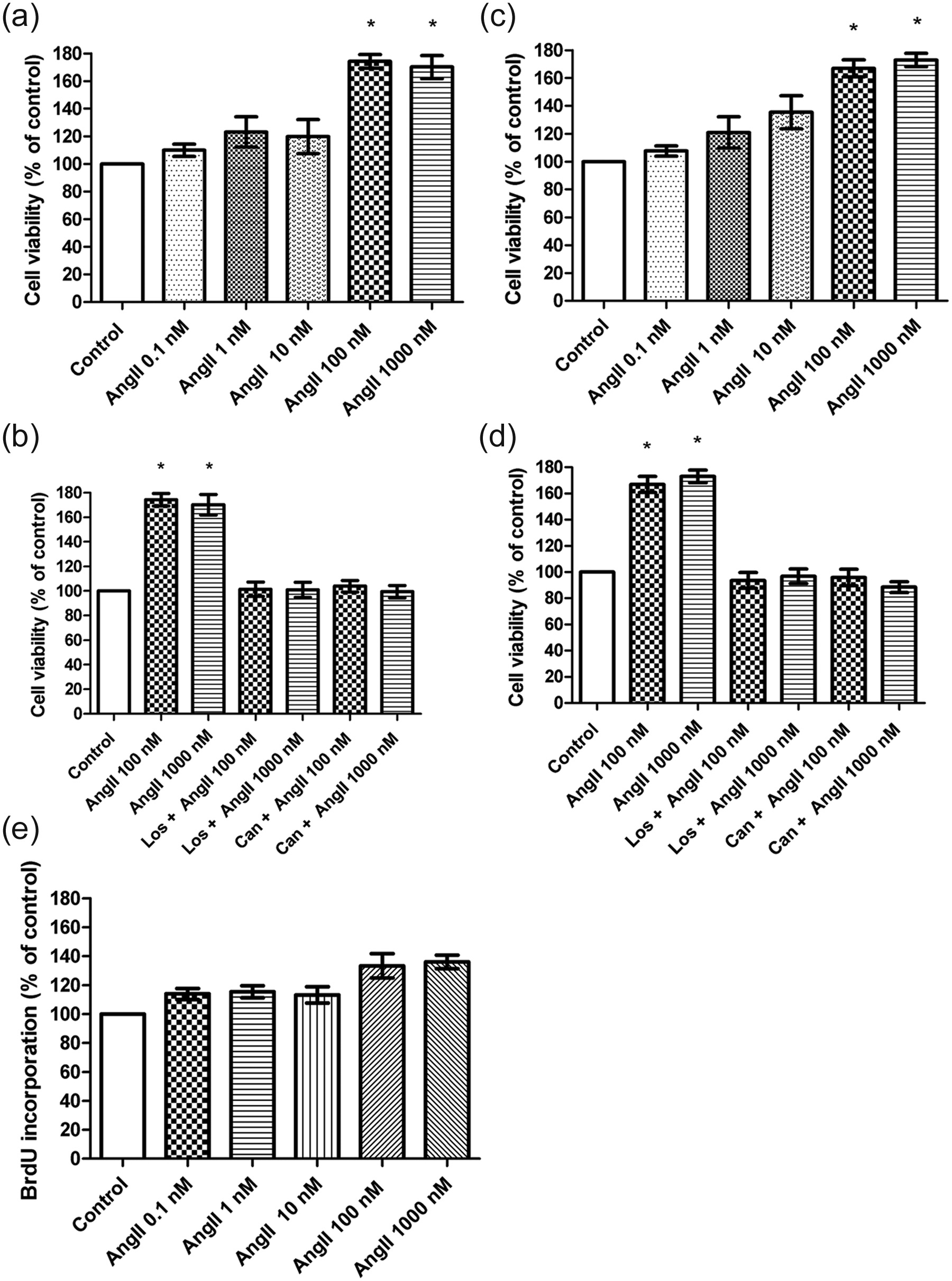
Effects of Angiotensin II on viability and proliferation of human mammary epithelial cells. Cell viability was measured using WST-1 and AlamarBlue assays. (a), (c) Cells were grown for 12 h (a) or 24 h (c) in the presence or absence of angiotensin II (AngII; 0.1 nM, 1 nM, 10 nM, 100 nM and 1000 nM); results are expressed as the percentage ratio over control cells. (b), (d) Effects of the AT1 inhibitors candesartan (Can) and losartan (Los) on the Ang II-stimulated (100 nM and 1000 nM) viability of 184A1 cells; cells were grown for 12 h (b) or 24 h (d) in the absence or presence of 1 µM candesartan or 1 µM losartan added 1 h before the administration of 1000 nM AngII; results are expressed as the percentage ratio over non-treated cells; bars show the mean ± SEM of three independent experiments performed in triplicate. *p < 0.05. (e) Cell proliferation measured by BrdU incorporation to cellular DNA; cells were grown for 24 h in the presence or absence of AngII (0.1 nM, 1 nM, 10 nM, 100 nM and 1000 nM); results are expressed as the percentage ratio over control cells. All results are representative for three separate experiments.
To evaluate the rate of cell proliferation in response to AngII, 184A1 cells were treated with 0.1 nM, 1 nM, 10 nM, 100 nM and 1000 nM) for 12 and 24 h. Only AngII (100 nM and 1000 nM) after 24 h was able to promote BrdU incorporation into DNA during the S-phase of the cell cycle (35.5 ± 16.4%; 36.8 ± 14.9%; p>0.05) (Figure 1(e)).
In order to evaluate whether the AT1-R are involved in AngII-induced cell viability and proliferation, 184A1 cells were treated with AT1-R antagonists candesartan and losartan for 1 h prior to the AngII treatment. Candesartan significantly suppressed AngII-induced (100 nM and 1000 nM) increase of cell viability at both time points 12 (Figure 1(b)) and 24 h (p ≤ 0.05) (Figure 1(d)). The second AT1-R antagonist, losartan, had similar efficacy to candesartan (p ≤ 0.05) (Figure 1(d)). Pretreatment with both inhibitors in combination was as effective as AT1 antagonist alone in modulation of the cell viability and proliferation. Candesartan or losartan treatment alone (without AngII) did not affect cell viability and proliferation.
On the basis of the results obtained at this stage of the research, in the next assays AngII was used at a concentration of 1000 nM, with 24 h incubation period with or without pretreatment with candesartan or losartan as AT1-R antagonists.
Cell adhesion
Treatment of 184A1 cells with AngII (1000 nM) for 24 h caused a slight reduction in adhesion to collagen type IV-coated (90% versus control) and laminin-coated (96% versus control) wells (Figure 2(a) and (b)). Further examination of the AngII treated cells showed that 184A1 cell adhesion was significantly increased (186% versus control) when using inserts coated with fibronectin (Figure 2(c) and (c′)). The effect of AngII on cell adhesion of 184A1 was attenuated by incubation with the AT1-R antagonists (Figure 2(a) to (c)).
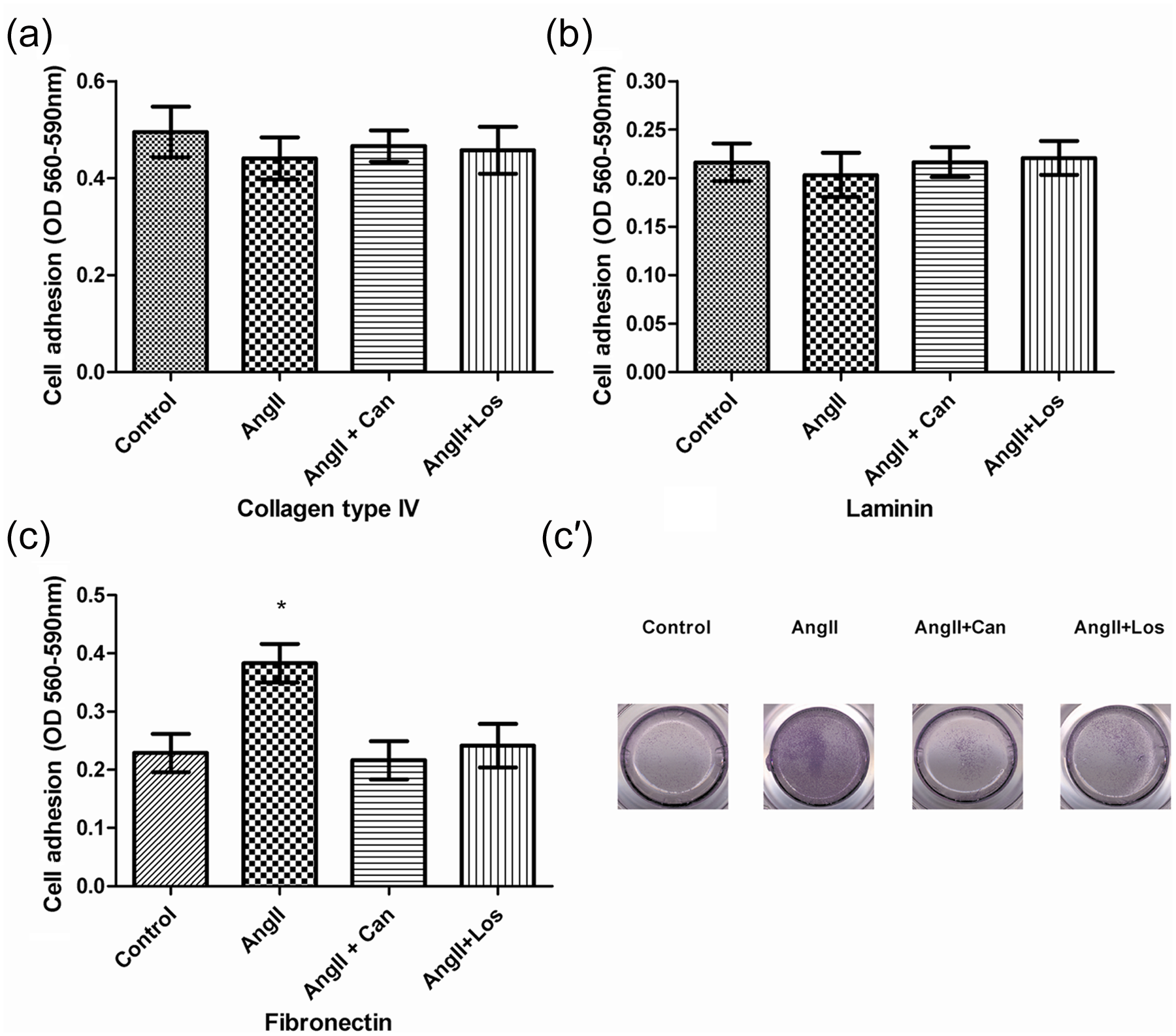
Angiotensin II (AngII) increases human breast epithelial cell adhesion. Adhesion of 184A1 human mammary epithelial cells to (a) collagen type IV-, (b) laminin- and (c) fibronectin-coated plates following exposure of 184A1 cells to AngII (1000 nM). Adhesive cells were stained with 0.1% crystal violet; the absorption was measured after lysis of the stained cells with 10% acetic acid. Results presented are mean ± SEM of three independent experiments performed in sextuplicate. *p < 0.05 compared with control. (c′) Photographs of cell adhesion to fibronectin-coated plates stained with 0.1% crystal violet.
Cell migration assays
Since our results have shown that AngII regulates cell adhesion, in the next step of the study we investigated the influence of AngII on motility. Treatment of mammary epithelial cells with AngII (1000nM) for 24 h significantly increased (1.2-fold) the ability of 184A1 cells to migrate in a Boyden chamber (Figure 3(a) and (a′)). Simultaneous pretreatment with candesartan or losartan abolished the effect of AngII on cell migration (Figure 3(a) and (a′)). Different results were obtained in invasion assays using inserts coated with GeltrexTM, which mimics the ECM. AngII (1000nM) was unable to increase the mammary epithelial cells’ invasiveness in a statistically significant manner at 24 h.

Angiotensin II (AngII) increases human mammary epithelial cells migration. Effect of AngII and AT1-R blockers on 184A1 cell migration. Modified Boyden chamber assays of human mammary cells migration across 8 µm pore non-coated membrane (a). Migratory cells were stained with 0.1% crystal violet, the absorption was measured after lysis of the cells with 10% acetic acid. Data are presented as the mean ± SEM of the absorption from three experiments performed in triplicate. *p < 0.05 compared with control. (a′) Photographs of cell migration through membrane stained with 0.1% crystal violet. (b) (b′) Wound healing assay; cells were plated onto individual wells of a six-well dish and allowed to grow to 80% confluence. A scratch was made across the centre of the cell monolayer with a yellow pipette tip creating a parallel space; chart (b) shows the results of three separate experiments performed in triplicate (mean ± SEM) and expressed as relative changes compared with control. *p < 0.05. Panel (b′) shows comparative microscopic view of 184A1cells in the scratched area at time 0 and after 24 h of a typical experiment.
The promigratory effect of AngII on breast epithelial cells was further confirmed in wound healing assays (Figure 3(b) and (b′)) showing significant increase (220% versus control) in cell migration and wound closure at 24 h following treatment with AngII (1000 nM). This migratory effect of AngII (1000 nM) was suppressed by candesartan to the level observed in the control group. The migratory abilities of cells induced by AngII were also inhibited in the presence of losartan, reaching almost the control values.
Gelatin zymography
Gelatin zymography revealed that 184A1 cells secreted into the culture medium proteins showing gelatinase activity, with a molecular mass of 72-kDa and 94-kDa after 24 h of incubation, which indicates that mainly MMP-2 and MMP-9 were secreted. AngII (1000 nM) stimulated MMP-2 and MMP-9 secretion and activity in 184A1 mammary epithelial cells (Figure 4(a) and (b)). Quantification of zymographic lysis bands indicated that this effect became statistically significant (p < 0.05) only in the case of MMP-2 (1.3-fold). Treatment with either candesartan or losartan suppressed AngII-induced MMP-2 secretion and activity (Figure 4(a) and (b)). Quantification of zymographic activity by densitometry indicated that this effect became statistically significant (p < 0.05).
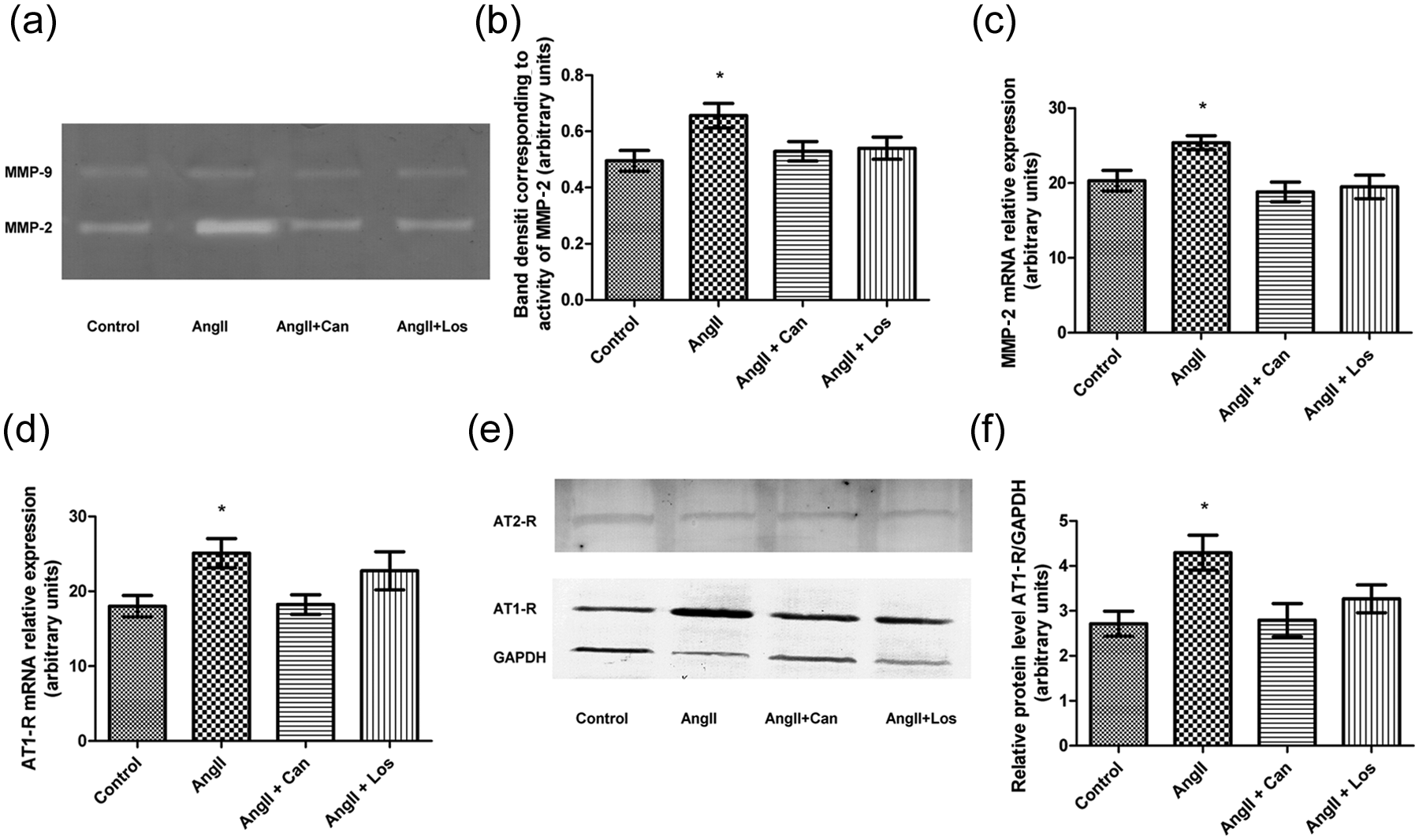
Effect of angiotensin II (AngII; 1000 nM) on activity and expression of MMP-2 in human mammary epithelial cells. (a) Gelatin zymography analysis of serum-free media conditioned by 184A1 cells treated with AngII. (b) Quantification of the gelatin zymography assay; *p < 0.05. Data are presented as the mean ± SEM of three separate experiments. (c) Real-time reverse transcription-PCR (RT-PCR) analysis of MMP-2 expression in 184A1 cells treated with AngII; results are mean ± SEM of three independent experiments performed in triplicate. (d) Real-time RT-PCR analysis of AT1-R expression in 184A1 cells pre-incubated for 1 h with candesartan (Can; 1 µM ) or losartan (Los; 1 µM) and treated with AngII for 24 h; bars show the mean ± SEM of three independent experiments performed in triplicate; *p < 0.05. (e) Western blot analysis of AT1-R protein level in 184A1 cells treated with AngII for 24 h. (f) Integrated optical density of the western blot assay; bars show the mean ± SEM of three independent experiments performed in triplicate; *p < 0.05.
Gene expression analysis/western blot analysis
First, we examined whether AT1-R and AT2-R mRNA and protein were expressed in 184A1 cells. The results of real-time RT-PCR indicate that only the expression of AT1R mRNA in the 184A1 cells treated with AngII was on a higher level in comparison with the non-treated cells (25.1 ± 1.4, 18.0 ± 1.9; p ≤ 0.05). Pre-treatment with candesartan, but not losartan, significantly decreased the expression of AT1-R gene when compared with the angiotensin treated cells (25.1 ± 1.4, 18.0 ± 1.3; p ≤0.05) (Figure 4(d)).
Protein expression of AT1-R and AT2-R showed a similar pattern to the results obtained in real-time RT-PCR. The results shown in Figure 4(e) and (f) indicate that the expression of AT1-R but not AT2-R protein in the 184A1 cells treated with AngII was higher than in the control (4.3 ± 0.4, 2.7 ± 0.3; p ≤ 0.05). This effect of AngII was significantly abolished when cells were pre-treated with candesartan (4.3 ± 0.4, 2.8 ± 0.4; p ≤0.05), but not with losartan.
In order to determine whether there is a relationship between the expression of angiotensin receptor genes and the effect of AngII on cell migration and invasion, the expression of two MMPs with known function in mammary gland physiology were also investigated. Only MMP-2 mRNA expression was induced by AngII. Treatment with candesartan or losartan suppressed this increase of MMP-2 expression in 184A1 cells (Figure 4(c)).
Discussion
In the normal cycle of changes in the mammary gland, the ductal system begins its elongation in puberty, stabilizes in adult life and develops extensively during pregnancy, enabling a high level of secretory activity during lactation. 25 The lactation cycle comprises a sequence of development and involution of the secretory tissue, accompanied by a dynamic balance between the synthesis and proteolysis of extracellular matrix proteins and other basement membrane components.2,25 The microenvironment of the mammary gland during these changes shares similarities with inflammation. 2
In many tissues, AngII plays a key role in remodelling, affecting both the activity of MMPs and cell mobility. 25 AngII is better known for being a central peptide in the regulation of systemic blood pressure and water and electrolyte homeostasis. Current studies point to an additional, separate, paracrine RAS in many tissues, including secretory epithelium of human mammary gland. 26 AngII is believed to allow tissue-specific function in the absence of more systemically released hormones, which could result in inappropriate or non-specific actions. 27 Evidence for this has been provided by in vitro studies demonstrating the changes in growth of various cells treated with angiotensin, angiotensin receptor antagonists or angiotensin converting enzyme (ACE) inhibitors.11,15 All components of RAS were found in most of the examined breast tissue samples. 25 Moreover, the AT1-R has been shown to be present in both human benign and human malignant breast cancer tissues. 28 The particular role of the AT1-R in human mammary epithelial cells is at present unclear, but it is downregulated in primary breast tumours. 26
In the present study we demonstrated that AngII was able to increase significantly the viability of 184A1 human mammary epithelial cells through mitochondrial metabolic activity, but it triggered the DNA synthesis (measured by the incorporation of BrdU) only moderately. Contrary to our results Greco et al. observed that the actions of AngII stimulated the proliferation of human primary mammary epithelial cells through signalling pathways mediated by the AT1 receptor. 29 This discrepancy could be dictated by different methods used for the evaluation of cell proliferation. In the case of our studies the highest mitochondrial activity measured in WST-1 and AlamarBlue assays (comparable to MTT) was treated as an indicator of cell viability, but not cell proliferation. Only incorporation of BrdU to new cells was recognized as the factor indicating new cells being created through mitotic division. The mitogenic effect of AngII was also demonstrated by Zhao et al. on MCF-7 breast cancer cells. 30 These authors also showed that AngII significantly enhanced cell-cycle progression by promoting the entry into S phase in MCF-7 cells. 30 In order to determine whether the AT1 receptor is responsible for mediating the proliferative effects of AngII, we used receptor specific inhibitors. Both AT1-R blockers candesartan and losartan suppressed the proliferative effects of AngII. These findings confirmed that AngII mediates cell proliferation through activation of AT1-R. The obtained results are consistent with previous reports showing that AT1-R antagonist suppressed the proliferative effects of AngII in MCF-7 cells. 30
Tahmasebi et al., using quantitative RT-PCR analysis, revealed the presence of AT1 and AT2 receptor mRNA in normal and pathological human breast tissue samples; however, the expression of AT1 gene was much more abundant in carcinomas than in normal tissue. 28 Our results showed that AngII was able to increase the AT1-R expression in human mammary epithelial cells. Moreover, this upregulation was connected with high cell viability and motility. Our observations confirmed that the stimulatory effect of AngII on cell viability and proliferation is mediated by AT1, which may have a key role in regulation of MMP-2 activity in 184A1 cells. These findings support the hypothesis indicating that the increased AT1-R expression may contribute to remodelling of the mammary gland’s microenvironment.25,28
We also investigated the role of AngII in cell substrate adhesion, migration and MMP secretion, which may be associated with tissue remodelling. Our study showed that AngII selectively increased cell substrate adhesion to a fibronectin-coated surface, improved wound closure, and was able to increase cell migration through non-coated membrane in Boyden chambers. Furthermore, AngII increased wound cluster, but did not affect the potency of cell migration through Geltrex-coated membrane. Deryugina et al. suggested that activation of MMP-2 and its proteolytic activity localized to the cell surface could differentially modulate cell migration in response to particular matrix proteins by altering both composition of the extracellular matrix and expression of adhesion receptors on the cell surface. 31 Furthermore Brooks et al. have shown that MMP-2 does not bind to the fibronectin receptor, suggesting that the MMP-2 is not able to affect the cells–fibronectin adhesion. MMP-2 is an important protease involved in normal and pathological tissue remodelling. In this study, we found that AngII increased the secretion of MMP-2 by mammary epithelial cells, while AT1-R antagonists were able to decrease this secretion. These observations may indicate that AngII, which affects cells via AT1-R, could induce matrix turnover by enhancing the activity to secrete MMP-2. 32 Similar results were presented by Rodrigues-Ferreira et al., who showed that AngII upregulates MMP-2 but not MMP-9 gene expression and enzymatic activity in mammary epithelial cell line MDA-MB-231, as well as in the gastric cancer cell line MNK-28. 7 Some interesting results were obtained by Nahmod et al., who showed that exogenous administration of AngII in mammary glands of lactating Balb/c mice induced epithelium apoptosis, and the effect was inhibited by irbesartan, an AT1-R blocker. These results, contrary to our observation, could be dictated by the use of a different research model. Moreover, this group has shown that AT1-R is involved in inhibition of MMP-9 activity during postlactational regression. All these observations strongly suggest that AngII, via the AT1-R, plays an important role in mouse mammary gland involution. The results reported by the cited authors together with our present study show how the RAS can play different roles depending on the stage of mammary gland development and differentiation. 33
Puddefoot et al. demonstrated that treatment of MCF-7 cells with AngII for 48 h caused concentration-dependent reduction in adhesion to fibronectin, laminin and collagen type IV-coated inserts. This significant inhibition of MCF-7 cell adhesion to fibronectin, laminin and collagen type IV by AngII was attenuated by incubation with the AT1-R antagonist losartan. 3 These observations are contrary to our results, and may indicate divergent effects of AngII in normal and cancer cells.
Coordination between remodelling of the ECM and changes in the receptors responsible for cell adhesion provide unique mechanisms regulating directional cell motility. In this context, MMPs, which potentiate ECM turnover, and molecules mediating cell adhesion to ECM proteins should combine effectively to facilitate cell migration and invasion under normal and pathological conditions. 31
Conclusion
The present study shows for the first time that direct exposure of human mammary epithelial cells to AngII creates changes in cell behaviour. Exposition of 184A1 cells on AngII caused overexpression of AT1-R but not AT2-R. Moreover, the alteration in the local RAS influenced cell viability and motility. Based on the presented data, we propose that changes in 184A1 cells contribute to the remodelling of the mammary gland. Our data support the hypothesis that AngII has a potency to modify cell behaviour and viability.
Footnotes
Acknowledgements
We would like to thank Mr Rafał Kasprzak and Mr Daniel Kasprzak for their support.
Conflict of interest
The authors declare no conflict of interest.
Funding
This work was supported by the Ministry of Science and Higher Education (grant number N403 293 539) and the Medical University of Lodz (grant number 502-03/0-078-04/502-04-008).