
Abstract
Introduction:
Angiotensin (Ang) A was first identified in human plasma and it differs from Ang II in Ala1 instead of Asp1. Here, we hypothesized that the actions of this peptide might explain, at least partially, the limited effects of AT1R antagonists in certain cardiovascular diseases.
Materials and methods:
The effects of Ang A and Ang II on blood pressure (BP) and heart function were compared. Importantly, participation of AT1R in these effects was evaluated. Furthermore, the effects of these two peptides on ischemia/reperfusion arrhythmias and involvement of calcium in these effects were investigated.
Results:
Administration of increasing doses of these peptides caused elevations in BP at comparable magnitude. AT1R blockade completely abolished these effects. The actions of these peptides in cardiac function were quite similar although the effects of Ang A were only partially blocked by losartan. Interestingly, Ang II elicited an increase in the duration of ischemia/reperfusion arrhythmias while Ang A had no effect on cardiac rhythm during reperfusion. In accordance, differently to Ang II, Ang A did not induce any significant effect on calcium transient during baseline and ischemic stress conditions.
Conclusions:
These data suggest that the existence of alternative peptides of the renin–angiotensin system (RAS) might contribute to the limited effects of angiotensin receptor blockers (ARBs) in certain pathophysiological circumstances.
Keywords
Introduction
Nowadays, one person in three suffers from some form of cardiovascular disease. This is the leading health problem in USA and cause of death. 1 Thus, there is an urgent need to expand our knowledge about this kind of disease, as well as develop new therapeutic strategies to improve the outcomes of cardiovascular disease.
The renin–angiotensin system (RAS) is a pivotal regulator of cardiovascular function and is over-activated in several diseases involving different organs, such as brain, lung, kidney and heart. The classical effector of this system, the octapeptide angiotensin (Ang) II, exerts its effects through specific G protein-coupled receptors (GPCRs) named AT1 and AT2.2,3 Under physiological and pathological conditions, it is well recognized that AT1 receptors play a critical role in the Ang II-mediated cardiovascular actions. 4 Thus, activation of this receptor is related to the promotion of vasoconstriction, inflammation, increases in blood pressure and cardiac dysfunction.5,6 Indeed, heart dysfunction has been associated with an increase in the expression of AT1 receptors in the myocardium immediately after ischemia-reperfusion without changes in the expression of AT2 receptors. Furthermore, these effects were attenuated by the specific AT1 receptor blocker losartan.7–9
The relevance of the RAS is highlighted by the success obtained in therapeutic strategies based on the pharmacological inhibition of this system using angiotensin-converting enzyme (ACE) inhibitors (ACEi) or angiotensin receptor blockers (ARBs) in cardiovascular diseases. Nevertheless, in some circumstances the effects of these drugs are limited. 10 In addition, there is recent evidence showing that part of the beneficial effects of ACEi and ARBs is mediated by increases in plasma Ang-(1–7) levels and ACE2 expression observed with the use of these drugs.11–13 Ang-(1–7), a heptapeptide of the RAS, and ACE2 hold several beneficial cardiovascular effects. 14 Therefore, these findings suggest that other active components of the RAS might exert crucial roles in the pathophysiology of the cardiovascular system.
In 2007, Jankowisk and co-authors described a novel peptide of the RAS named Ang A. 15 This peptide was observed in human plasma and its sequence of amino acids is composed by Ala-Arg-Val-Tyr-Ile-His-Pro-Phe. Therefore, Ang A differs from Ang II in Ala1 instead of Asp1, i.e. Des[Asp1]-[Ala1]-Ang II. Ang A is synthesized by enzymatic decarboxylation of Asp1 of the Ang II sequence. In isolated perfused rat kidney, Ang A caused a vasoconstrictive effect dependent on AT1 receptors. 15 Furthermore, it has been reported that Ang A elicited pressor and renal vasoconstrictor responses in normotensive and hypertensive rats which were abolished by candesartan, an AT1 receptor antagonist. 16 However, the role of Ang A in the cardiovascular system, especially in the pathophysiology of the heart, is still poorly explored. Thus, in the present study, we focused on two major objectives: (a) to evaluate the role of Ang A in the pathophysiology of the heart and (b) to test the hypothesis that the actions of this peptide might explain, at least in part, the limited effects of ARBs in some cardiovascular diseases. To achieve these objectives we investigated the effects of Ang A on blood pressure, cardiac function and calcium handling in rats, as well as the role of AT1 receptors in these effects.
Materials and methods
Chemicals and reagents
Ang II was obtained from Bachem (Bachem, Torrance, California, USA) and losartan was purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, Missouri, USA). Ang A was prepared on an ABI model 431A automated peptide synthesizer using the software version 1.00 for Fmoc synthesis (Applied Biosystems, Carlsbad, California, USA).
Animals
This study was conducted in male Wistar rats (200–300 g) from CEBIO - Federal University of Minas Gerais (Belo Horizonte, MG, Brazil). The animals were housed in a temperature-controlled room (22–23°C) with a 12–12 h light-dark cycle. Water and food were available ad libitum. All experimental protocols were performed in accordance with the Federal University of Minas Gerais (Brazil) Institutional Animal Care and Use Committee, which is in compliance with the National Institutes of Health (NIH) guidelines.
Blood pressure measurements
Under anesthesia with 12% urethane [1 ml/100 g of body weight, intraperitoneal (i.p.)], a catheter was inserted into the carotid artery and jugular vein for blood pressure measurement and drug injections, respectively (n=6). The arterial catheter was connected to a strain-gauge transducer coupled to a computer-based data acquisition system (MP100A, Biopac Systems Inc., California, USA) to record pulsate arterial pressure. The animals were maintained under anesthesia and were warmed with a heated pad. The mean arterial pressure (MAP) was simultaneously calculated by the Acknowledge software (Biopac Systems Inc., California, USA) and displayed continuously. After an initial 30 min stabilization period, increasing doses of Ang II or Ang A (12.5, 25, 50, 100 and 200 ng/kg in ~0.1 ml of saline) were given intravenously. To evaluate the role of AT1 receptors in the effects of Ang A on MAP, the animals were pre-treated with losartan (5 mg/kg) 30 min before Ang A injections.
Isolated heart preparation
Additional groups of rats (n=6−8) were heparinized (400 IU, i.p.), decapitated and the hearts were rapidly excised and cooled in ice-cold Krebs-Ringer solution (KRS) (118.40 mM NaCl; 4.70 mM KCl; 1.17 mM KH2PO4; 1.17 mM MgSO4; 2.50 mM CaCl2; 11.65 mM glucose and 26.30 mM NaHCO3) to preserve the heart before the perfusion. The hearts were perfused through an aortic stump with KRS at 37±1°C in a Langendorff system with constant pressure (65 mmHg) and oxygenation (5% CO2 and 95% O2). A force transducer (TSD 104 A, Biopac Systems, Inc., California, USA) was attached through a heart clip to the apex of the ventricles to record the contractile force using a data-acquisition system (Acknowledge, Biopac Systems, Inc., California, USA). A diastolic tension of 1.0±0.2 g was applied to the hearts. Coronary flow was measured by collecting the perfusate over a period of 1 min at regular intervals. Heart rate (HR) was calculated from the contractile tension recordings. The rate of tension development (±dT/dt) was obtained by calculating the derivatives using the Acknowledge Software (Biopac Systems, Inc., California, USA). Maximum dT/dt (+dT/dt) is a reasonable index of the velocity of myocardial contraction and minimum dT/dt (–dT/dt) is an index of the velocity of myocardial relaxation. After 30 min of stabilization, the hearts were perfused with KRS alone (control) or KRS containing Ang II (70 nM), Ang A (70 nM) or Ang A plus losartan (2.2 µM) for an additional period of 30 min. After this period, the left anterior descending coronary artery (LAD) was ligated beneath the left auricular appendage together with the adjacent veins. The ligature was released after 15 min, and reperfusion was performed for an additional 30 min. Cardiac arrhythmias were defined as the presence of ventricular tachycardia and/or ventricular fibrillation after the ligature of the coronary artery was released. To obtain a quantitative measurement, the arrhythmias were graded arbitrarily by their duration. Therefore, the occurrence of cardiac arrhythmias for up to 3 min was assigned the factor 2; 3–6 min was assigned the factor 4; 6–10 min was assigned the factor 6; 10–15 min was assigned the factor 8; 15–20 min was assigned the factor 10; 20–25 min was assigned the factor 11; and 25–30 min was assigned the factor 12. A value of 0–12 was thus obtained in each experiment and was denoted as the arrhythmia severity index (ASI). 17 The dose of Ang II and Ang A was based on a previous study. 18
Calcium recordings
Left ventricular cardiomyocytes were isolated, as described previously. 19 Briefly, hearts were quickly removed, mounted and perfused using a customized Langendorff apparatus for 5 min with nominally Ca2+-free solution containing (in mM): 130 NaCl, 5.4 KCl, 0.5 MgCl2, 0.33 NaH2PO4, 3 pyruvate, 22 glucose and 25 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH set at 7.4. Next, hearts were perfused for 10–15 min at 37°C with a solution containing 1 mg/ml type II collagenase (Worthington Biochemical Co, Freehold, New Jersey, USA). The left ventricular free wall was separated, minced, gently agitated, centrifuged (500 rpm, 1 min, room temperature) and stored in Tyrode solution. Only calcium-tolerant, quiescent, rod-shaped myocytes showing clear cross striations were studied. Intracellular Ca2+ imaging experiments were performed with Fluo-4 AM (10 μM; Invitrogen, Eugene, Oregon, USA)-loaded cardiomyocytes for 35 min, and these were subsequently washed with Tyrode solution to remove the excess dye. Cells were electrically stimulated at 1 Hz to produce steady-state conditions. The confocal line-scan imaging was performed with a Zeiss LSM 510META confocal microscope (Centro de Aquisição e Processamento de Imagens do ICB/UFMG). The Ca2+ level was reported as F/F0, where F0 is the resting Ca2+ fluorescence.
Low concentrations of a glycolysis inhibitor (0.5 mM 2-deoxy-D-glucose) and of an oxygen scavenger (0.5 mM sodium dithionite, Na2S2O4) were diluted in a glucose-free Tyrode (CIS) solution to induce acute and mild cellular ischemic stress. 20 The cardiomyocytes were superfused over laminin-treated glass slides with Tyrode solution for 10 min followed by 4 min of mild metabolic inhibition and anoxia (perfusion with CIS solution) and then re-superfused with Tyrode solution for an additional 5 min. To evaluate the role of Ang II and Ang A in the amplitude of electrically-induced [Ca2+]i transient, the peptides (200 nM) were added to the Tyrode and CIS solutions. The electrically-induced [Ca2+]i transient was evaluated after (a) superfusion (baseline), (b) CIS solution perfusion and (c) reperfusion of the same cell with Tyrode solution (n=8). Pilot experiments were performed in order to determine the lowest dose of Ang II and Ang A able to induce consistent results. Thus, after testing the doses of 70, 100 and 200 nM, we chose the dose of 200 nM.
Statistical analysis
Statistical analysis was performed using the unpaired Student’s t-test, one-way analysis of variance (ANOVA) followed by the Newman-Keuls post-hoc test or two-way ANOVA followed by the Bonferroni post-hoc test. Differences were assumed to be significant for values of p<0.05.
Results
Effects of Ang A on MAP
Ang A elicited a dose-dependent increase in the MAP of rats. These actions were similar to the pressor effects of Ang II (100 ng/kg: Ang II 13.15±4.00 mmHg and Ang A 12.30±3.00 mmHg; Figure 1(A)). Moreover, these pressor responses were mediated by AT1 receptors since co-treatment with losartan completely blunted the effects of Ang A (Figure 1(B)). As expected, a similar blockage was observed when the animals were administered with Ang II plus losartan, indicating that the concentration of losartan utilized was sufficient to block the AT1 receptors (Figure 1(C)).
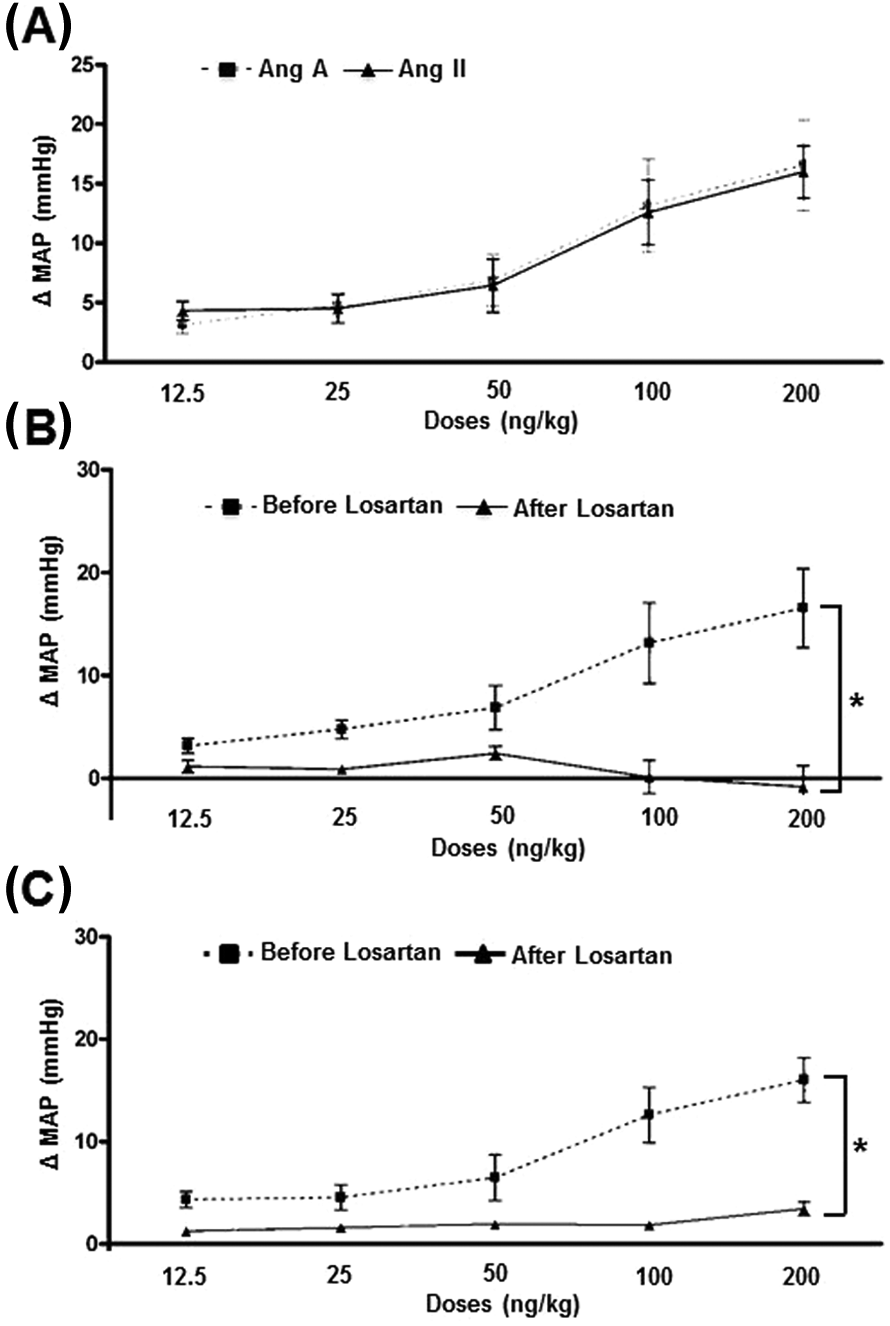
Effects of angiotensin (Ang) A on mean arterial pressure (MAP) of rats. (A) Ang A and Ang II elicited similar increases in MAP. (B) and (C) These effects were completely abolished by losartan.
Effects of Ang A on cardiac function
To evaluate the cardiac effects of Ang A, we used an isolated heart preparation. Infusion of Ang A caused a significant reduction in the coronary flow, +dT/dt, –dT/dt and HR when compared with hearts perfused with KRS alone (Figures 2(A)–(D)). Although Ang II induced similar effects on these parameters, we observed a statistical difference only in coronary flow when compared with control hearts (Figure 2(A)). Of note, the effects of Ang A on cardiac function were partially blocked by losartan, demonstrating that AT1 receptors participate in the Ang A actions in hearts (Figures 2(A)–(D). No significant changes were observed in systolic and diastolic tension among any of the groups (Figures 2(E) and 2(F)).

Effects of angiotensin (Ang) A on cardiac function of isolated hearts. (A) Coronary flow (ml/min); (B) +dT/dt (g/s); (C) –dT/dt (g/s); (D) heart rate (HR), (bpm); (E) systolic pressure (g); and (F) diastolic pressure (g) of hearts perfused with Krebs-Ringer Solution (KRS) alone (control) or KRS containing Ang II (70 nM), Ang A (70 nM) or Ang A plus losartan.
In terms of cardiac arrhythmias, the presence of Ang II in the perfusion solution provoked a significant increase in the duration of ventricular tachycardia and/or ventricular fibrillation during the reperfusion of the myocardium after ischemia (Figure 3(A)). However, even though perfusion of Ang A was also able to cause an increase in the severity of the cardiac arrhythmias, it did not reach statistical significance (Figure 3(B)).

Effects of angiotensin (Ang) A on ischemia/reperfusion arrhythmias in isolated hearts. Perfusion of (A) Ang II, but (B) not of Ang A, caused a significant increase in the arrhythmia severity index (ASI).
Effects of Ang A on cardiac calcium handling
In order to establish the participation of intracellular calcium in the actions of Ang A in cardiac contraction and reperfusion arrhythmias, we carried out experiments utilizing isolated ventricular myocytes. Acute and mild cellular ischemic stress induced by CIS solution caused no cell death (data not shown) and increased the magnitude of electrically-induced [Ca2+]i transient after reperfusion of cardiomyocytes (Figure 4(A)). Furthermore, it was observed that the peak of the electrically-induced [Ca2+]i transient was increased by Ang II during the baseline and CIS perfusion conditions when compared with ventricular myocytes perfused with Tyrode solution alone. On the other hand, Ang A did not elicit any significant effect on electrically-induced [Ca2+]i transient during the baseline, CIS perfusion and reperfusion conditions. No significant differences were observed among any of the groups in the reperfusion period (Figure 4(B)).

Effects of angiotensin (Ang) A on calcium handling of isolated cardiomyocytes. (A) Effects of mild cellular ischemic stress induced by glucose-free Tyrode solution containing 2-deoxy-D-glucose and sodium dithionite (CIS) and reperfusion on electrically-induced [Ca2+]i transient in left ventricular myocytes. (B) Left ventricular myocytes superfused with Tyrode (control), Ang II (200 nM) or Ang A (200 nM) during baseline, CIS solution perfusion and reperfusion. *p<0.05 compared with baseline in (A) and with Tyrode group in (B). One-way analysis of variance (ANOVA) followed by the Newman-Keuls post-hoc test.
Discussion
The main findings of the present study are that Ang A, a peptide produced by decarboxylation of Ang II, is a biologically active member of the RAS with many actions similar to Ang II. Specifically, these actions include elevation of blood pressure, coronary vasoconstriction and reduction in cardiac contraction and HR. Moreover, we found that AT1 receptors are, at least in part, involved in the Ang A effects. On the other hand, in contrast to Ang II, this peptide did not increase reperfusion arrhythmias and change the amplitude of the calcium transient in isolated cardiomyocytes.
The first evidence for the pathophysiological relevance of Ang A came from its identification in human plasma of healthy subjects and of patients with end-stage renal failure. Importantly, it was shown that these patients presented a higher ratio between Ang A and Ang II. Furthermore, a vasoconstrictive effect of Ang A was described using an isolated perfused rat kidney preparation. 15 In line with these findings, we found that Ang A has many cardiovascular actions. Altogether, these data evidence the complexity of the RAS, indicating that new components of this system might play a crucial role in the pathophysiology of the cardiovascular system.
We found that the hypertensive effect of Ang A is dependent on AT1 receptors since losartan was able to completely blunt this action. Indeed, Yang et al. reported that Ang A elicited pressor and renal vasoconstrictor responses in normotensive and hypertensive rats, which were abolished by candesartan, an AT1 receptor antagonist. 16 On the other hand, our data demonstrated that the cardiac effects of Ang A were only partially mediated by binding of this peptide to AT1 receptors. Therefore, to our best knowledge, this is the first study showing that the role of AT1 receptors in the effects of Ang II and Ang A may differ depending on the organ evaluated. This might explain, at least in part, the limited actions of ARBs in some conditions as Ang A had similar actions to Ang II effects and AT1 blockade resulted in distinct actions in the cardiac Ang A effects. In fact, it has been reported that the vasoconstrictive effects of Ang A in isolated perfused rat kidney are smaller when compared to the Ang II actions, thereby suggesting a lower intrinsic activity at the AT1 receptors in this vascular bed. 15 Altogether these findings suggest the existence of a distinct receptor for Ang A.
Among the cardiovascular parameters evaluated in this present study, the effect of Ang A on cardiac arrhythmias was one of the most distinct actions when compared to Ang II. In fact, while Ang II caused a significant enhancement in the duration of the reperfusion arrhythmias, Ang A induced only a slight increase in this parameter. In addition, because Ca+2 handling is one of the major determinants of the incidence and duration of cardiac arrhythmias, we evaluated the magnitude of the Ca+2 transient in isolated cardiomyocytes subjected to acute and mild cellular ischemic stress induced by CIS solution. Accordingly, only cardiomyocytes perfused with Ang II presented an increase in [Ca2+]i transient during baseline and CIS perfusion. Evidence shows that the RAS is activated during ischemia and reperfusion, with the Ang II peptide being one of the main causes of the cardiac damage.21–23 Reperfusion arrhythmias are associated with changes in Ca+2 levels and it is well established that Ang II not only stimulates Ca+2 influx through Ca+2 channels but also induces release of intracellular Ca+2, resulting in excess of Ca+2 inside the cells during ischemia/reperfusion injury. 8
Conclusions
In summary, our findings showed that Ang A is a cardiovascular active peptide of the RAS, further indicating that the complexity of this system is beyond that which we have thought previously. The effects of this peptide were not identical to those of Ang II. Furthermore, the blockade of Ang II and Ang A effects by losartan was distinct. Altogether, these data provide evidence that the existence of alternative peptides in the RAS, such as Ang A, might contribute to the limited effects of ARBs in certain pathophysiological circumstances.
Footnotes
Conflict of interest
No potential conflicts of interest were reported.
Funding
This work was supported in part by FAPEMIG-Brazil (Fundação de Amparo à Pesquisa do Estado de Minas Gerais), CNPq-Brazil (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and CAPES-Brazil (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).