
Abstract
Tiotropium Bromide is a long-acting bronchodilator that is used in the treatment of chronic obstructive pulmonary disease (COPD) and asthma bronchodilator or bronchiolitis, which are substances that expand the bronchi and reduce resistance in the respiratory tract and increase airflow to the lungs. For Tiotropium Bromide found in inhaler capsules to treat COPD, determining the relevant impurities G and H, which are not UV active, is crucial. For this purpose, a new and sensitive liquid chromatography triple-quadrupole mass spectrometry (LC-MS/MS) detection with electrospray ionization by using multiple reaction monitoring in the positive mode method was developed and validated. The identity of the compounds was supported by using LC-Q/TOF. All chromatographic studies were performed with a Zorbax Eclipse XDB-C8 (150 mm x 4.6 mm, 5.0 µm) column with a total injection time of 13 min at a flow rate of 0.4 ml/min as a gradient. The limit of detection (LOD) and limit of quantitation (LOQ) in the current study range were determined as 1.0 ppb and 2.5 ppb, respectively. The results of the validation parameters following the ICH Q2(R1) guideline were determined within the acceptance criteria.
Introduction
Tiotropium bromide (Figure 1) is an anticholinergic agent with long-acting, which is allowed to be used in many countries for use in the treatment of patients with chronic obstructive pulmonary disease. It is thought that the reason for this long effect in the body is that Tiotropium Bromide blocks the M1, M2, and M3 subtypes of muscarinic receptors in the airway.1–3 Also defined as a bronchodilator or muscarinic antagonist, Tiotropium Bromide widens the bronchi to allow more airflow to the lungs and reduces resistance in the respiratory tract.4,5 Tiotropium Bromide, which has a long duration of action in metabolism, is sufficient to use once a day by inhalation for treatment due to this feature.4,6–8
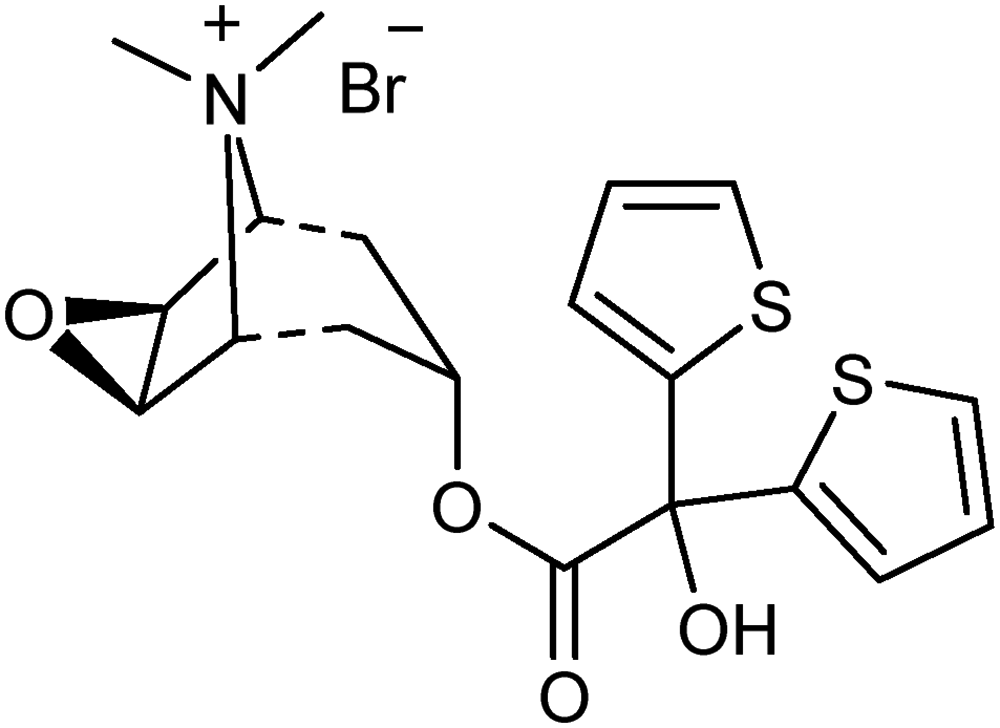
Chemical structure of tiotropium bromide.
There are many analytical methods for the determination of Tiotropium Bromide, which is applied by dry powder inhalation and is present in very low concentrations in drug formulations. When the British (BP) and European (EP) pharmacopeias are examined, it should be noted that there is only Tiotropium Bromide raw material monograph.9,10 A, C, E, and F impurities (Figure 2) are controlled by the same HPLC method in both pharmacopeias, and it is seen that the same TLC method is given for G and H impurities. For the assay method, the potentiometric titration method is given. In the literature research, it is observed that there are studies including different analytical techniques, mostly assay. RP-HPLC systems used in inhaler products6–8,11 and liquid chromatography triple-quadrupole (LC-MS) methods used especially for determinations in human plasma1,12 are generally studied for the Tiotropium Bromide assay. In the literature research conducted for the determination of Tiotropium Bromide impurities, no methods other than RP-HPLC were found, and it was seen that known impurities were not defined in these studies. Therefore, the studies were carried out on unknown and total impurities.13,14
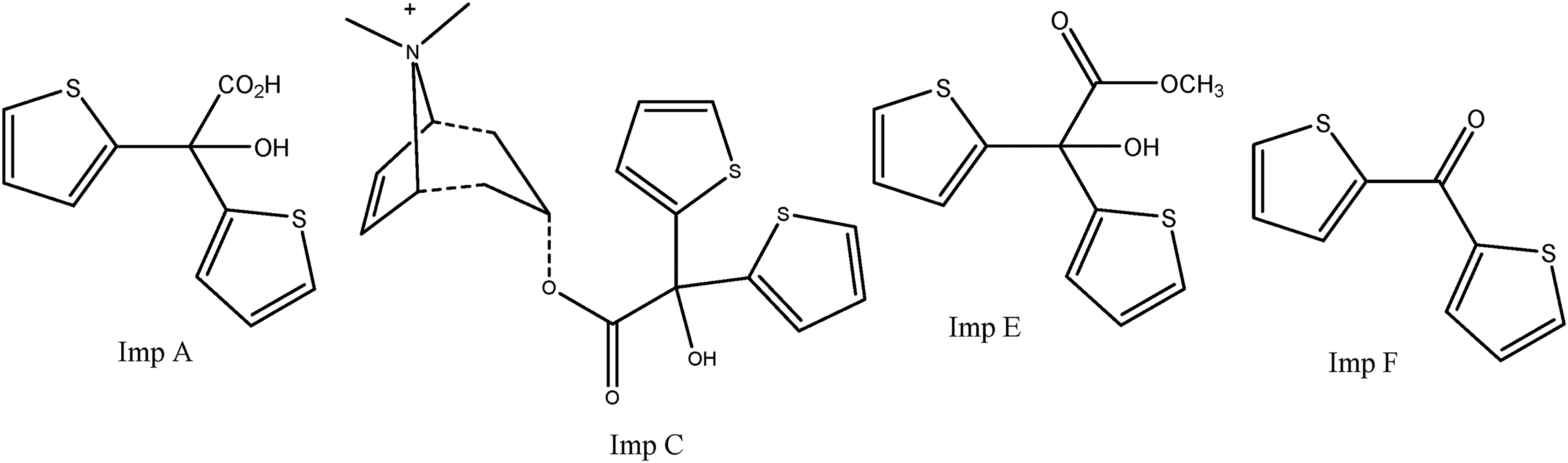
Structures of tiotropium bromide impurities A, C, E, and F.
According to The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q3B(R2) guideline, known and unknown impurity limits for drug products are determined based on the daily dose of the product. 15 Known and unknown impurity limits for drug products containing Tiotropium bromide at a level of µg as a label value are quite low, and it is very difficult to determine specific impurities such as impurity G and impurity H, which are not UV active (Figure 3). In EP and BP monographs, the impurity G and H limits required to be controlled is 0.1%, depending on the calculation made on the daily dose for Tiotropium Bromide raw material, while this limit drops to 18 ppb (µg/L) in finished product forms. Therefore, it is very difficult to determine such low-impurity concentrations by chromatographic techniques such as TLC. At this point, since the monograph method is not applicable, a new method of research and development was carried out to determine the impurity G and H. Based on this requirement, the study proposes a new and sensitive method of mass spectrometry with high-resolution liquid chromatography triple-quadrupole mass spectrometry (LC-MS/MS) for impurities G and H. In addition, the formation of the impurities obtained by stress test studies was supported by characterizing them with LC-Q/TOF.
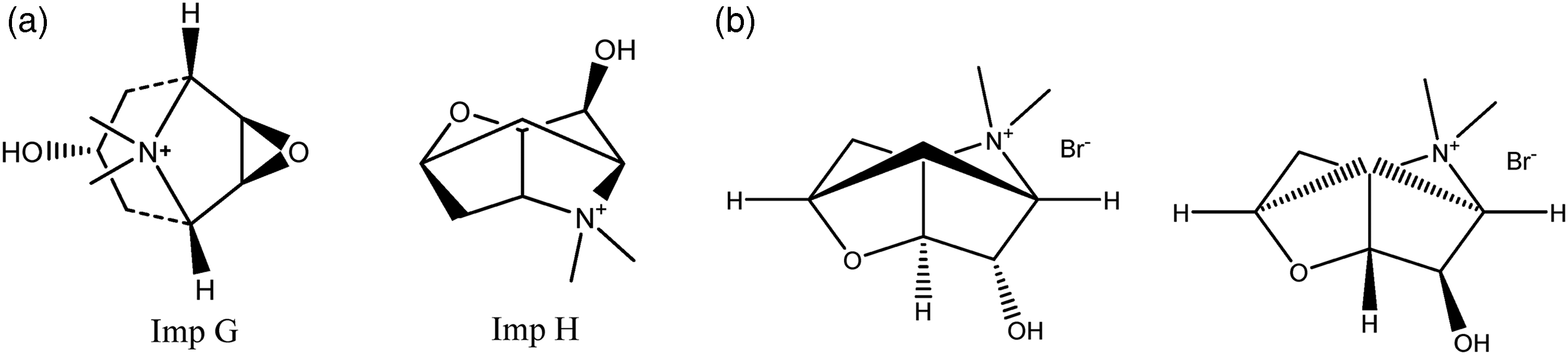
(a) Structures of tiotropium bromide impurities G and H. (b) Two enantiomeric forms of impurity H depending on the epoxide opening side.
Materials and pharmaceutical sample
Tiotropium bromide monohydrate from EP and impurity G and impurity H standards from Simson Pharma (Dahisar, Mumbai) were used as standards in the studies. To prepare the mobile phase, LC-MS grade acetonitrile, water, and ammonium formate were purchased from Merck (Darmstadt, Germany) and formic acid (98%) was obtained from Thermo Fisher (Waltham, MA, USA). A 0.45 µm PTFE filter was produced from Sartorius (Göttingen, Germany).
The inhaled capsule containing Tiotropium Bromide as the active substance used in the studies was produced by World Medicine Ilac San. ve Tic. A.S (Istanbul, Turkey).
Instrumentation
This method used for quantification was developed and validated on the Agilent 6470A Triple Quadrupole LC/MS (Agilent, USA) equipped with a Dual AJS electrospray ionization (ESI) ion source consisting of LC-1260 Infinity II HPLC system (Agilent, USA). Data acquisition and analysis were performed using Mass hunter 10.1 software.
The chromatographic separation was performed on the Zorbax Eclipse XDB-C8 column (150 × 4.6 mm, 5 µm) with gradient elution at a flow rate of 0.4 ml/min. The mobile phase-A was 0.1% formic acid containing 5 mM ammonium formate solution and mobile phase-B was LC-MS grade acetonitrile. The gradient program was created as follows: 0–1 min, 10% B; 1–9 min, 10% →80% B (linear); 9–11 min 80% B; 11–11.1 min 80% →10% B(linear); 11.1–13 min using 10% B. The flow rate was 0.4 ml/min and the total run time was 13 min. The autosampler and column oven temperatures were 25°C and 30°C, respectively. Compounds were detected using multiple reaction monitoring (MRM) in positive mode. Optimized operating source conditions for MS scanning in positive-ion ESI mode: the capillary voltage 3500 V; nitrogen was used as the drying (300°C; 6 L/min) and nebulizer (45 psi) gas. The mass spectra were recorded across the range of m/z 50–750 for both positive and negative ion modes. Transitions of m/z 170 > 84.1 (collision energy: 36) and m/z 170 > 72.1 (collision energy: 46) and m/z 170 > 58.1 (collision energy: 44) were used for the analysis of tiotropium impurity G and H. Given the distinct matrix interference of the sample, m/z 170 > 84.1, m/z 170 > 72.1 and m/z 170 > 58.1 were used for quantitative analysis of Tiotropium bromide impurity G and H in inhaler capsules.
To scan for impurity G and H degradation impurities in stress studies, a liquid chromatography quadrupole time-of-flight mass spectrometer (Agilent Q-TOF-MS 6546) is equipped with an ESI source of a 1290 series HPLC instrument (Agilent Technologies, USA) was used. The column, flow rate, mobile phases, and gradient program were kept the same as LC-MS/MS. The typical operating source conditions for MS scan in positive and negative ion ESI modes were optimized; the capillary voltage was 3500 V; the skimmer at 65 V; nitrogen was used as the drying (320°C; 8 L/min) and nebulizer (35 psi) gas. The mass spectra were recorded across the range of m/z 100–3000 for both positive and negative ion modes. Nitrogen was kept as a nebulizer and auxiliary gas. The data acquisition was carried out by Mass Hunter workstation software.
Method development
In the EP Tiotropium bromide, raw material monograph impurity G- and H-related compounds method, Tiotropium bromide active substance concentration was determined as 40 mg/ml. And the standard solutions prepared at all limit concentrations were calculated over this concentration. To bring the capsule samples containing 22.5 µg Tiotropium Bromide equivalent to 18 µg Tiotropium to the concentration indicated by the monograph, approximately 2000 capsules must be dissolved with 1 ml of solvent, which shows that this method is impossible considering the excipients in the product. However, sample preparation procedures at low concentrations have been tried for Tiotropium bromide and it has been observed that there are placebo-induced solubility problems in these studies. For this reason, an alternative to the TLC method given in the monograph, simpler and more selective method studies have been carried out for finished product formulations.
In the literature research, no method has been found for the determination of impurity G and H. Quantification methods, mostly RP-HPLC methods, have been encountered for Tiotropium, which is mostly found in different combined products. Quantitative studies of Tiotropium, which is found at very low concentrations in human plasmas, have been carried out by HPLC/MS/MS methods. 1 Method development studies were carried out with LC-MS for these two impurities, which were UV inactive and would give a sensitive response at ppb levels when compared to other impurities whose molecular shapes are given in Figure 3.
Method optimization
The mass spectrometer was set to scan the mass range of 50–500 m/z, due to the low molecular weights of impurity G and impurity H. In the scanning results, it was observed that both impurities gave a dominantly 170.0 m/z ion peak in positive mode, but impurity G gave 160.7 m/z and impurity H 160.8 m/z molecular ion peaks in negative mode. As the peak intensities were higher for the positive mode as seen in Figure 4, the positive-ion tracking mode in a triple-quadrupole HPLC-ESI-MS-MS was chosen for the studies. Positive mode scanning was expected to dominate for impurity G and impurity H already containing quaternary ammonium in their structure.
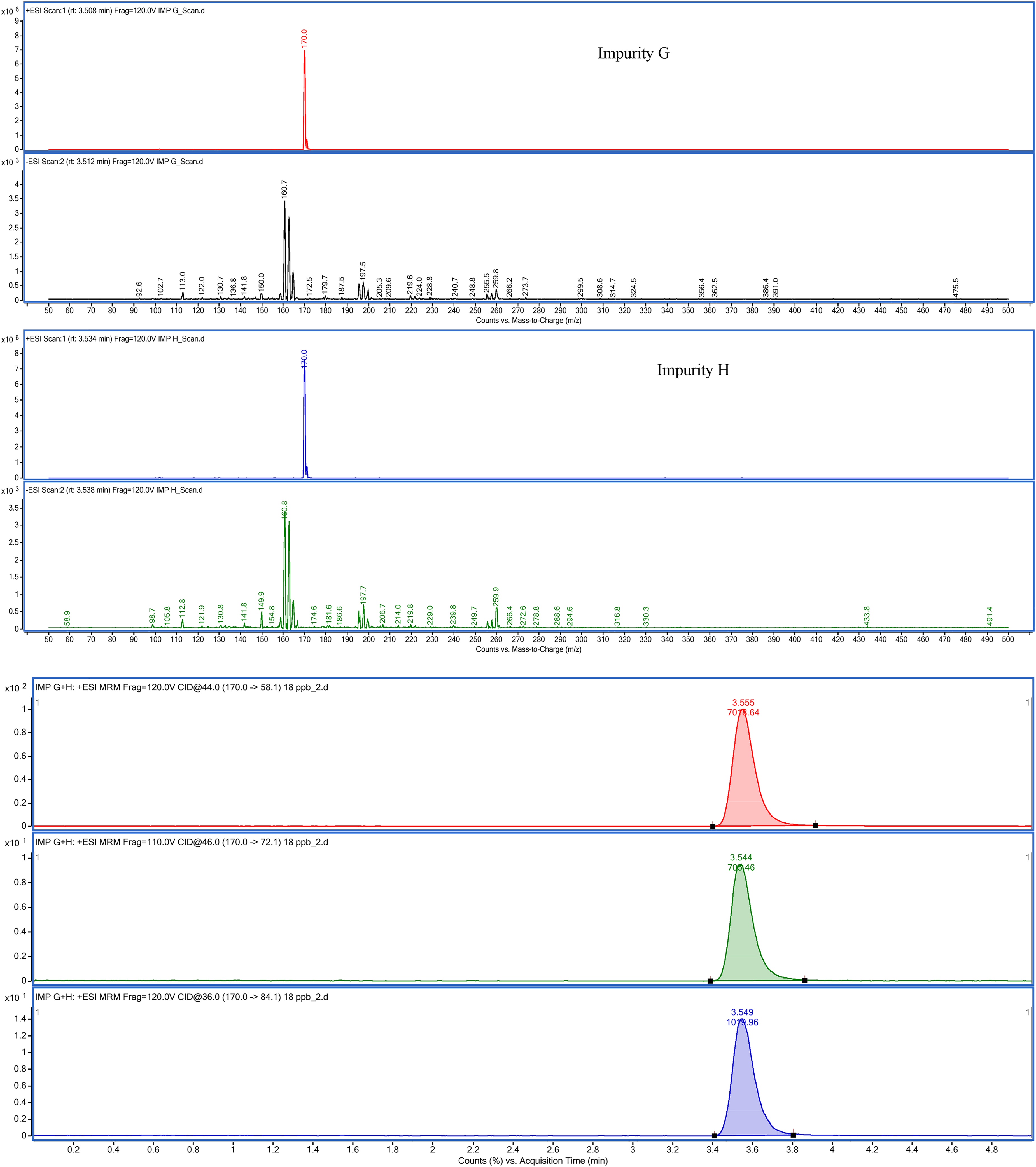
Mass range scan results for impurity G and impurity H and XIC of the precursor ion and the product ions.
To determine the optimum breakdown voltage, the intensity of impurity G and impurity H molecular ion [M]+ m/z 170.0 is 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, and 130 V were compared at fragmentor voltages. The highest sensitivity was found for both impurities at 120 V fragmentor voltage. In the MS/MS studies, the precursor ion (m/z 170.0) gave three prominent product ion peaks at m/z 84.1, m/z 72.1, and m/z 58.1, and these product ions were found to be common to both impurities. Since Impurity G and Impurity H are two structural isomers with the same closed formula, they gave the same product ions at the same retention time, the same fragmentor, and collision voltages for the same precursor ion. For this reason, it was not appropriate to evaluate these two impurities separately, and the impurity was controlled with a narrow limit of 1.0% as G + H.
In experiments with mobile phases containing different combinations of acetonitrile, methanol, ammonium acetate, ammonium formate, and water, it was observed that the presence of 5 mM ammonium formate containing 0.1% formic acid both increased the ionization and made the peak shapes sharper. In addition, the use of acetonitrile provided strong signals. In trials with several commercial columns such as ACE, Zorbax, Inertsil, Nucleosil, Hypersil, and Kromasil, the most satisfactory chromatography was obtained with the Zorbax Eclipse XDB-C8 column (150 × 4.6 mm, 5 µm) because of the pressure problems and the peak shapes.
Method validation
The analytical method developed specifically for the determination of Tiotropium Bromide impurities G and H in inhaler products has been validated following the ICH Q2 (R1) Validation of Analytical Procedures: Text and Methodology guideline and USP <736> Mass Spectrometry monograph.16,17
Linearity
Linearity is defined as an interval in the measurement range of an analytical method in which an output signal correlates linearly with the determined analyte concentration. By MRM, the linearity of impurity G and impurity H was satisfactorily demonstrated with a five-point calibration graph between 2.5 and 30.0 ppb with respect to a sample concentration of 1.8 ppm. Approximately 2 mg of impurities G and H were accurately weighed, transferred to a 10 ml volumetric flask, and dissolved and diluted with acetonitrile to volume. Fifty microliters of this solution was diluted to 10 ml with acetonitrile to obtain 1000 ppb stock standard solution. Using this stock impurities were gradually diluted to concentrations of 2.5, 10, 18, 25, and 30 ng/ml. The intercept, slope, and correlation coefficient were determined by linear regression data analysis and fitting to a linear regression model with a weighting scheme of 1/x. The regression equation and correlation coefficient with the MRM fields corresponding to the concentration levels are given in Table 1. In addition, Figure 5 shows that the standard curve consisting of Impurity G and Impurity H was linear in the concentration range of 2.5–30 ng/ml and the correlation coefficient (r) > 0.99 meets the acceptance criterion. The chromatogram and spectra of the linearity study are given in Supplementary Material Figure S-1—Figure S-5.
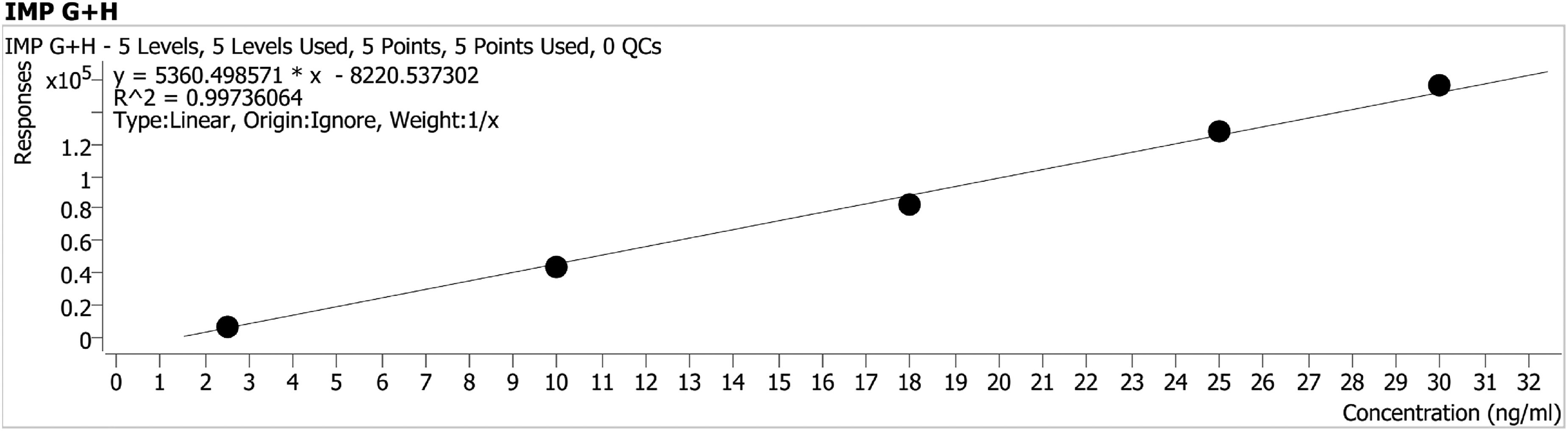
Calibration graph of impurity G + H.
Linearity results of the method.
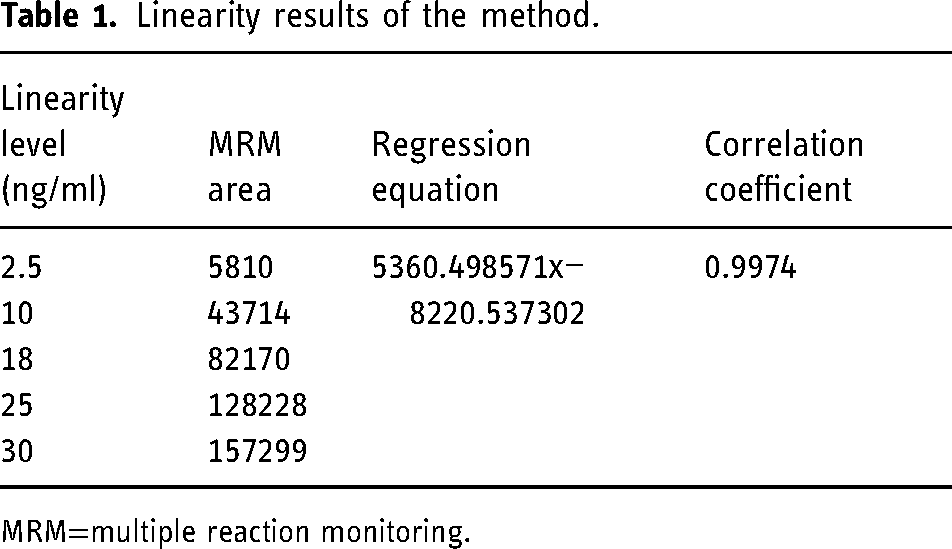
MRM=multiple reaction monitoring.
Under optimized conditions, the accuracy of the impurity G and impurity H components was evaluated by the recoveries. Typically, three levels of concentrations were evaluated. According to the USP <736> Mass Spectrometry monograph, the LOQ is verified by measuring six replicates of test samples spiked with analyte at 50% of specification. Considering this information, the accuracy of the method was determined by spiking at 9.0 ppb (LOQ), 18.0 ppb (specification limit), and 25.0 ppb (about 140% of the limit) concentrations separately to formulation solutions of tiotropium bromide (1.8 ppm). A total of nine samples including three for LOQ and three for other levels, were prepared and analyzed. Readings were made so that the LOQ solutions were six replicates and the other solutions were three replicates. According to the USP <736> Mass Spectrometry monograph; recovery should be between 70%—130% for LOQ and 80%—120% for other concentrations. In addition, the RSD value of the six measurement results given for the LOQ should be at most 15%. It was shown in Table 2 that the data obtained as a result of the study met the acceptance criteria. The chromatogram and spectra for accuracy and LOQ are given in Supplementary Material Figure S-6—Figure S-8.
Accuracy and LOQ results of the method.
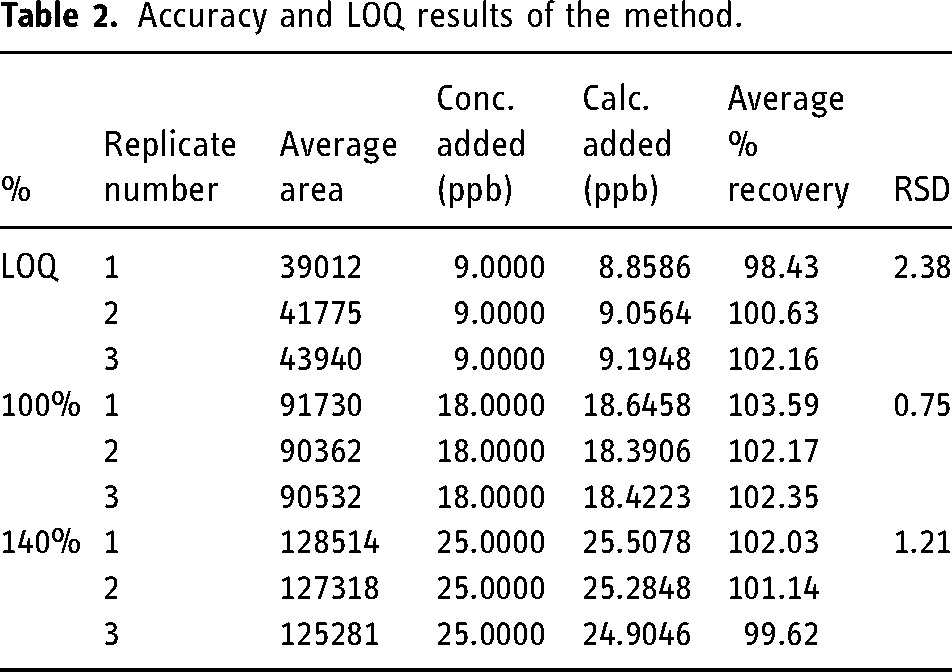
Accuracy and LOQ results of the method.
The precision and intermediate precision (on a different day by a different analyst) parameters were controlled with six different sample solutions spiked with impurity G + impurity H at the specification limit (18 ppb). Each solution was given to the system once. The results of the recovery of impurity G + impurity H were checked by calculating the RSD value. Acceptance criteria and results for both system precision and intermediate precision are given in Table 3. Representative MRM chromatograms are given in Figure S-9 and Figure S-10.
Precision and intermediate precision results of the method.
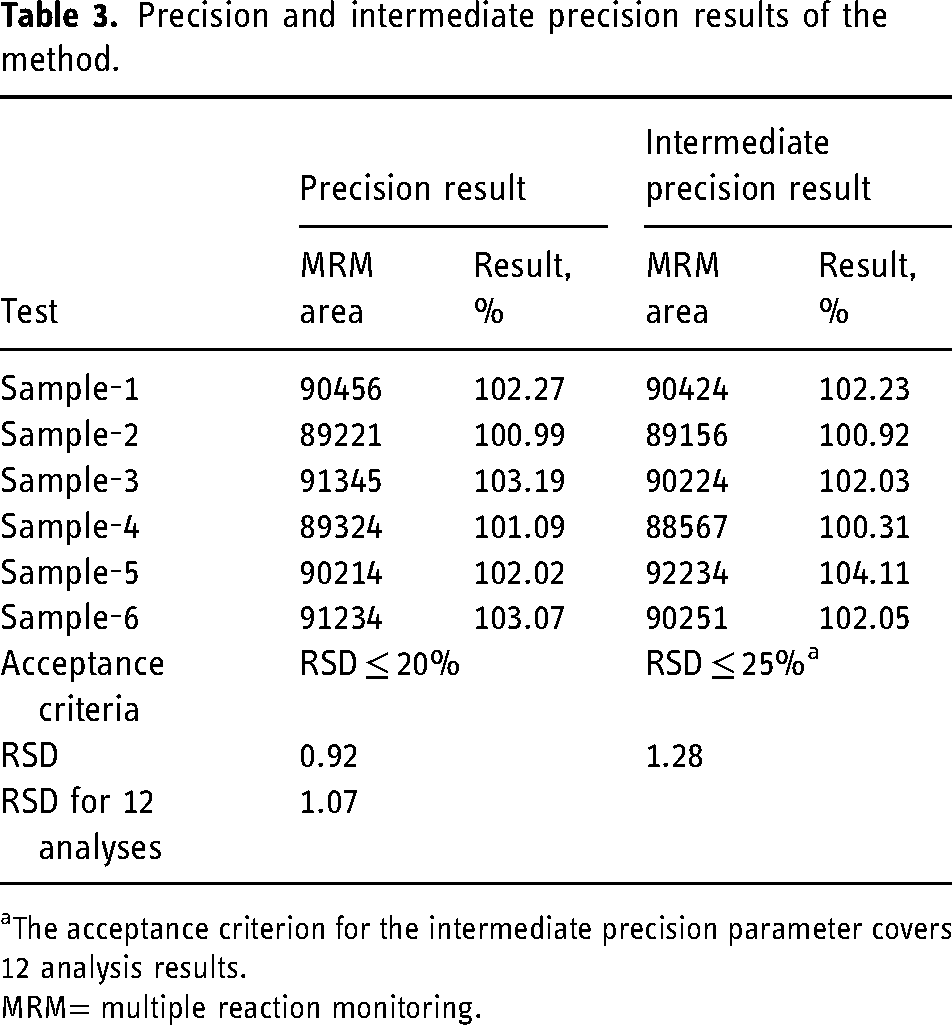
Precision and intermediate precision results of the method.
The acceptance criterion for the intermediate precision parameter covers 12 analysis results.
MRM= multiple reaction monitoring.
The robustness of the method was studied with deliberate modifications in the flow rate of the mobile phase and the column temperature. The optimized flow rate of the mobile phase was 0.4 ml/min and the flow rate was changed to ±0.1 ml/min while keeping the other parameters constant. The effect of column temperature on peak area was studied at 28°C and 32°C (altered by 2°C).
The stability of impurity G and impurity H in standard and sample solutions was checked by keeping them at room conditions and following the variations in their peak areas.
It was observed that the solutions were stable for 48 h in line with the comparisons made with the initial area value for the solution stability parameter. With the changes to make control the durability of the method, it was determined that there were no remarkable areal differences when compared with the initial value. As an acceptance criterion, it was evaluated whether the change from the initial value was greater than 20%. Representative MRM chromatograms are provided in Figure S-11 and Figure S-12.
Specificity
The specificity of the method was established by injecting, dilution solution, blank Tiotropium Bromide (capsule) solution, and impurity G and impurity H spiked at the specification limit. It was observed that the common excipients used in the capsules did not interfere with the retention times of impurity G and impurity H. The corresponding specificity chromatogram is shown in Figure 6.
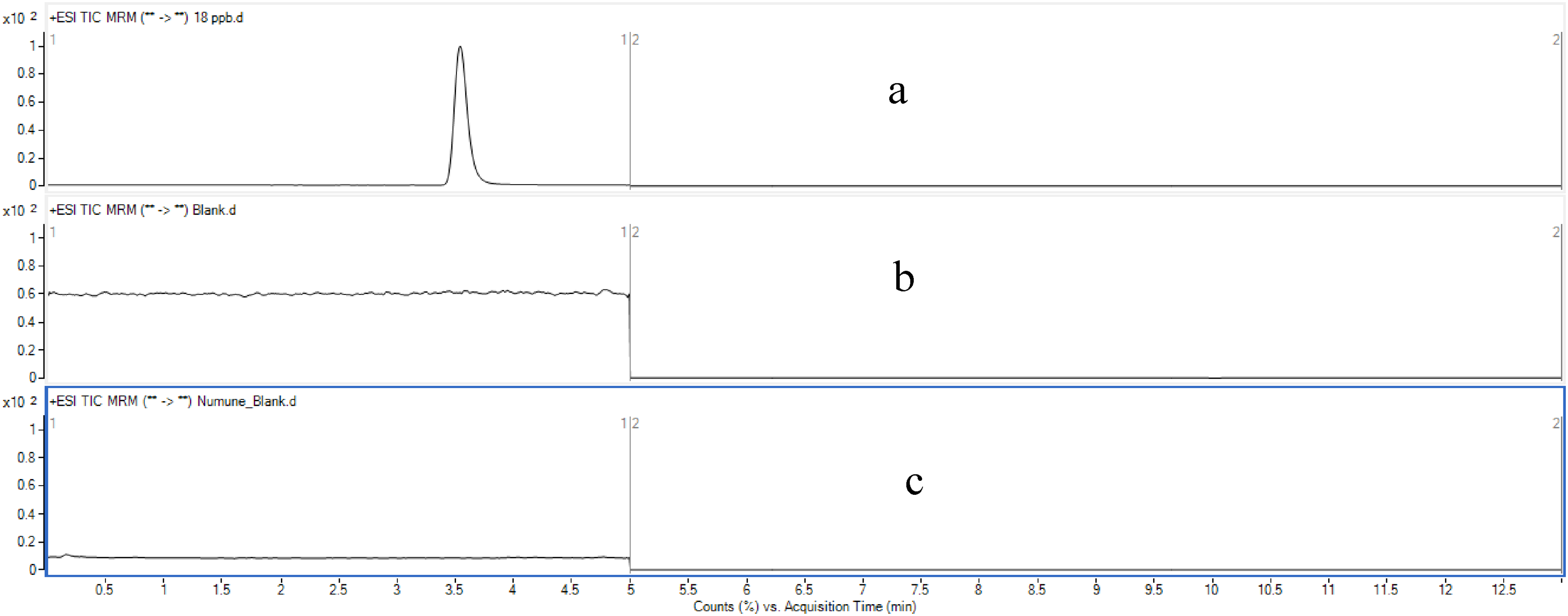
Specificity chromatograms—(a) sample solution spiked with impurity G and impurity H at the specification limit. (b) Dilution solution chromatogram (LC-MS Grade Acetonitrile). (c) Blank Tiotropium Bromide (capsule) solution.
Tiotropium Bromide raw material is easily converted to Impurity G by acidic or basic hydrolysis. To support this, the finished product was subjected to alkaline degradation (1 M NaOH, 25°C, 1 h) and injected into the LC-Q/TOF system. When the MS/MS spectrum was taken for the peak detected differently from the blank, the molecular ion peak was determined as 170.1187 m/z (Figure 7).
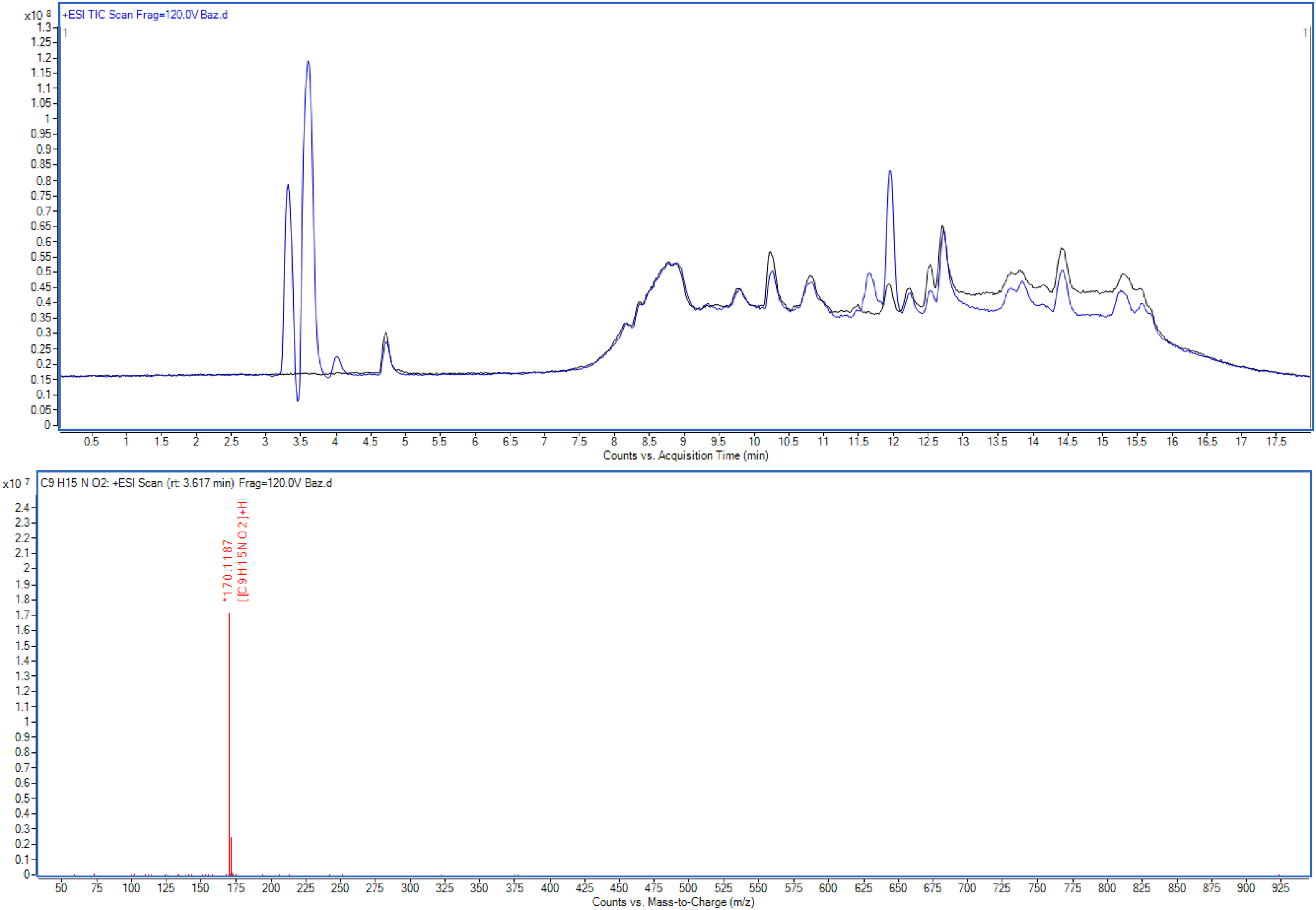
Chromatogram and MS/MS spectrum of base degradation.
The MassHunter Molecular Structure Correlator (MSC) software was used to determine the possible molecular structure of the detected ion. This software associates the correct mass MS/MS fragment ions for the compound in question with plausible substructures and confirms a proposed structure with higher confidence. The possible fragmentation structures suggested by the MSC software for the alkali degradation product are shown in Figure 8. As seen here, the structures belonging to the M/z = 170 ion formed by the fragmentation of the Tiotropium molecule are the dark marked part, and when the Tiotropium impurities are examined, it has been determined that the structure belongs to Impurity G. Similar results were obtained in acidic degradation studies for the finished product.
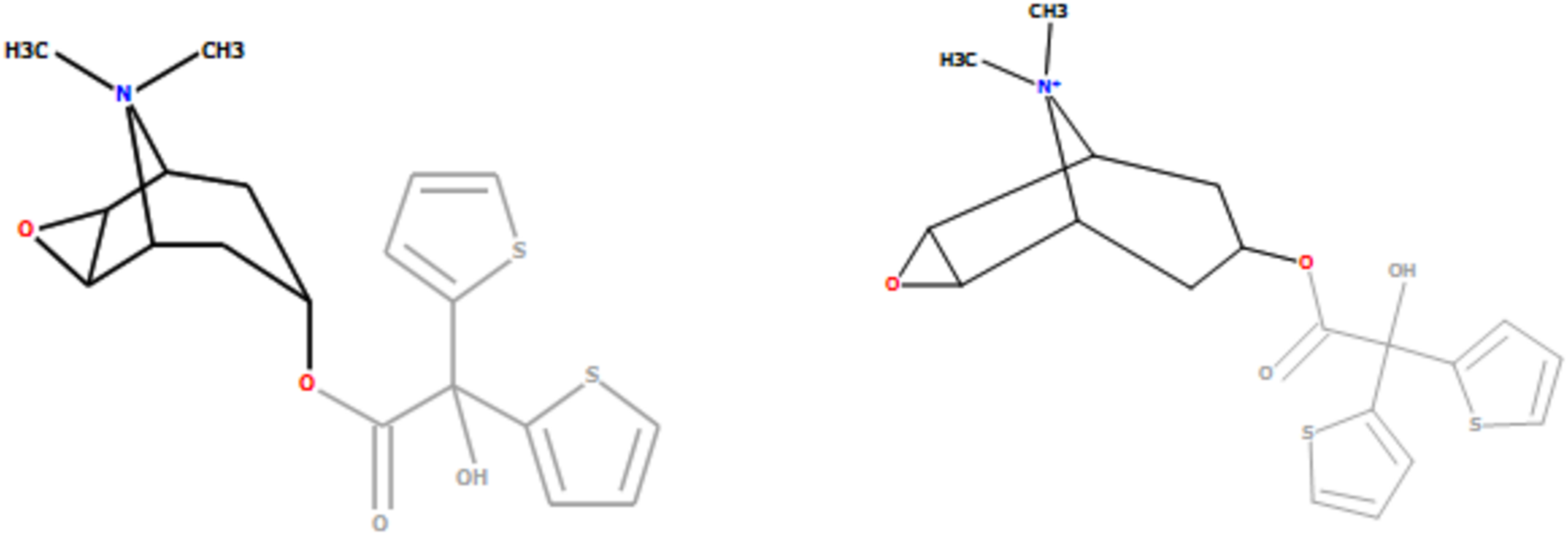
Possible structures for m/z = 170.
Since impurity H and impurity G are structural isomers of each other, it is expected that impurity H will be converted to impurity G by intramolecular cyclization. In the literature research, it is stated that this transformation occurs at a high temperature (90˚C). 18 The mechanism of conversion of Impurity G to Impurity H is given in Figure 9. Therefore, the finished product containing tiotropium was considered to decompose to Impurity G in acidic or alkaline decomposition conditions.
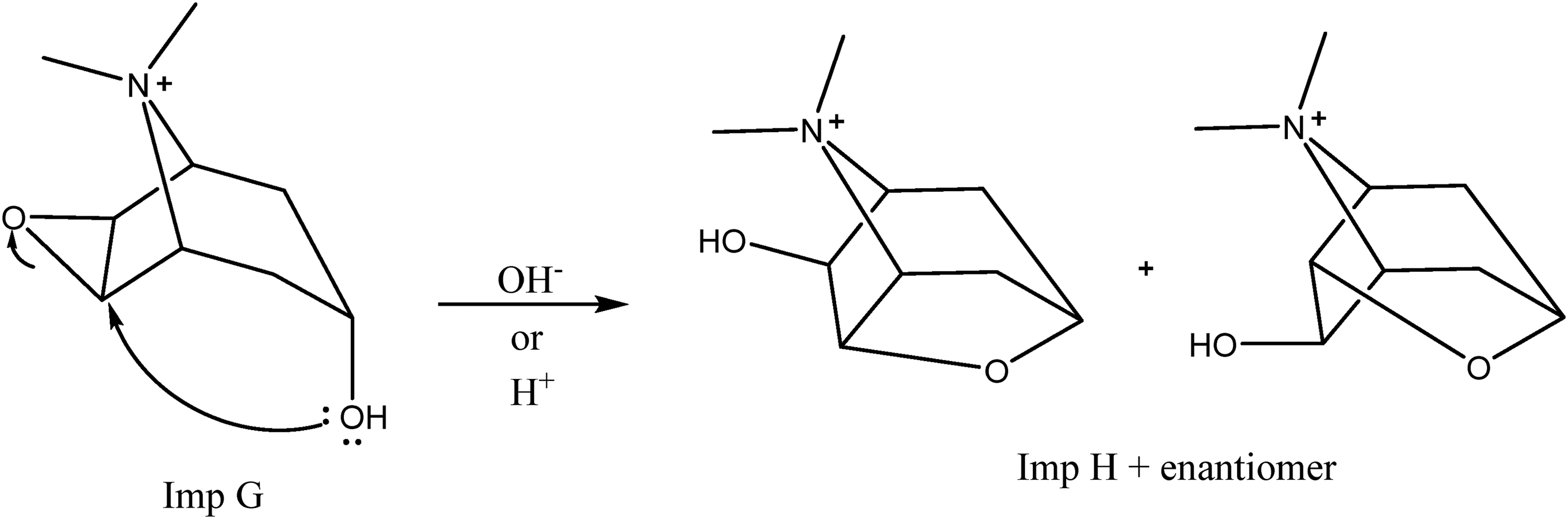
Mechanism of conversion of impurity G to impurity H.
It is necessary to develop sensitive and selective methods for the determination of impurities in finished products containing µg of an active substance. For this reason, a new LC-MS/MS method has been developed and validated for the determination of impurity G and impurity H within the limits determined according to the daily dose in accordance with the internationally accepted guidelines for the inhaler capsule product containing 18 µg Tiotropium. It has been explained in detail that the TLC method given in the EP and BP monographs is not suitable for these two impurities, which are not UV active and are structural isomers of each other, considering the amount of active ingredient and impurity limits in the finished product. There is no method in the literature research for these two impurities. It was necessary to develop a new method, especially in terms of routine quality control analysis. Since it is not possible to detect these two impurities separately even with a sensitive method such as mass spectrometry, evaluations were made as total impurities and studies have proven that the method is safe, linear, recoverable even at LOQ concentration, and precise even in a narrower limit than ICH limits.
Method development and validation studies were based on the USP <736> Mass Spectrometry guidelines together with the ICH Q2(R1) guidelines. Impurity G obtained by the degradation of Tiotropium Bromide was characterized by LC/Q-TOF, and its conversion to Impurity H was explained in detail. This method, which does not require a specific pre-treatment for product preparation, is quite simple and can be preferred for it is time-saving and economical with a total injection time of 13 min.
Supplemental Material
sj-docx-1-ems-10.1177_14690667231217879 - Supplemental material for Simultaneous quantification of tiotropium bromide impurities G + H in capsule formulation by LC-MS/MS
Supplemental material, sj-docx-1-ems-10.1177_14690667231217879 for Simultaneous quantification of tiotropium bromide impurities G + H in capsule formulation by LC-MS/MS by Serdar Erkol, Müge Güleli, Cem Çalışkan and Sevgi Kocaoba in European Journal of Mass Spectrometry
Footnotes
Acknowledgements
The authors are thankful to the management of World Medicine İlaç San. ve Tic. A.Ş (İstanbul, Turkey), for supporting this work.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.