
Abstract
The oxidation of furfural (2-furaldehyde), by benzimidazolium dichromate (BIDC) in dimethyl sulfoxide, leads to the formation of 2-furoic acid. The reaction is first order with respect to BIDC and hydrogen-ion. However, Michaelis–Menten type kinetics was observed with respect to furfural. The formation constants of furfural-BIDC complexes and the rates of their decomposition have been evaluated at different temperatures. Thermodynamic parameters of the complex formation and activation parameters for the decomposition of the complexes have been calculated. The deuterium isotope effect observed in the oxidation of furfural (kH/kD = 6.23 at 298 K) indicated an α-C-H bond cleavage in the rate-determining step. The reaction has been studied in 19 organic solvents. The analysis of the solvent effect showed that the role of cation-solvation is major. Based on the kinetic data, analysis of the solvent effect and the result of some non-kinetic parameters, a mechanism involving rate-determining oxidative decomposition of the complex, through hydride-ion transfer via a cyclic transition state, to give the corresponding product is suggested.
Introduction
In synthetic organic chemistry, selective oxidation of organic compounds under non-aqueous conditions is an important transformation. For this purpose a number of different chromium (VI) have been reported1–5. A new Cr(VI) derivative - benzimidazolium dichromate (BIDC) was reported in 1998 6 . This new compound is more efficient for quantitative oxidation of several organic substrates and has certain advantages over similar oxidizing agents in terms of the amount of oxidant and solvent required, short reaction time and high yields. Since BIDC is neither light sensitive nor hygroscopic, therefore, it is more stable and stored easily as compared to other Cr(VI) derivatives. It is reported 6 to convert allylic and benzylic alcohols to corresponding carbonyl compounds with 75–98% yield from BIDC.
We get interested in the kinetics and mechanistic study of the oxidation by BIDC and a few reports have already been emanated from our laboratory7–13. The kinetics of oxidation of furfural have been studied in presence of acid by many reagents such as 1-Bromobenzimidazole (BBI) 14 , Imidazolium dichromate (IDC) 9 , Quinolinium dichromate (QDC) 10 , Quinolinium chlorochromate (QCC) 20 , quinolinium flurochromate (QFC) 21 Nicotinic dichromate (NDC) 19 , Isonicotinium dichromate (INDC) 20 , Chromic acid 21 , N-bromonicotinamide (NBN) and N- Chloronicotinamide (NCN) 22 in binary mixture of acetic acid and water medium. In recent years some reports of oxidation of furfural over AuPd/Mg(OH)2 under basic conditions 23 and with hydrogen peroxide using sulfated zirconia 24 have been published. Furfural base-free oxidation over Au-Pd embedded bimetallic nanoparticles 25 , has also been reported.
In the oxidation of heteroaldehydes there exists clear feasibility of the reaction taking place either at the carbonyl function or at heteroatom. For the purpose of establish the site of reaction in the oxidation of heteroaldehyde, we have focused attention on oxidation of furfural and some substituted furfurals. A study of literature appears that there may be no report on the kinetic and mechanism of the oxidation of furfural by BIDC. In this paper, we report the oxidation kinetics of furfural by BIDC in dimethyl sulfoxide (DMSO) as solvent. Mechanistic aspects are discussed.
Experimental
Materials
BIDC was prepared by the reported method 6 and its purity was checked by an iodometric method. Furfural (Sigma-Aldrich) was distilled and the fraction collected at 162oC was used. Other substituted furfurals were also purified by distillation under reduced pressure. Deuterated furfural (furfural-D4) is available with high degree of purity and used as supplied. The solvents were purified by the reported methods 26 . Among the solvents, CS2 is a flammable liquid and toxic. Toluene p-sulphonic acid (TsOH) was used as a source of hydrogen ions. The solutions of TsOH were standardized by titrating them against standard alkali.
Product analysis
The oxidation of furfural leads to formation of 2-furoic acid. In this experiment, furfural (9.6 gm, 0.1 mol), TsOH (19.02 gm, 0.1 mol), and BIDC (4.54 gm, 0.01 mol) were composed to 100 ml in DMSO and it was then kept in the dark under nitrogen for a day to make sure the completion of reaction. The solution was rendered alkaline and evaporated to dryness under lower pressure. The residue was extracted three times with diethyl ether. The ether extract was dried with anhydrous Na2SO4. The product of oxidation was obtained after complete removal of ether. It was analysed by TLC, IR (KBr) and its melting point. The yield of the product formed was 84–89%.
Stoichiometry
Stoichiometry of the oxidation of furfural by BIDC.

Kinetic measurements
The pseudo-first-order conditions were carried out at constant temperature (±0.1) for all kinetic runs, by maintaining a large excess of the furfural (10 fold or greater) over BIDC. The reaction was carried out under DMSO solvent, unless specified. The progress of reaction was studied by monitoring the consumption of BIDC at 372 nm for up to 80% of the reaction. The Beer’s law is valid for all concentration range used in our experiments. The rate constant, kobs, was evaluated from the linear (r2 > 0.996) plots of log[BIDC] against time. The values observed were the mean of two or more than two runs (reproducibility ±3%). As the measures of the goodness of fit, we have used coefficient of determination (R2 or r2), standard deviation (SD) and Exner’s parameter 27 , ψ, in correlation analysis.
Results
The stoichiometric determination indicated the following overall reactions for the oxidation of furfural.
Test for free radicals
Rate constants for the oxidation of furfural by BIDC at 308 K [H+]= 1.0 mol dm−3.

ac (acrylonitrile) = 0.001 mol L−1.
bc (acrylonitrile) = 0.005 mol L−1.
Rate laws
The reactions were found to be of first order with respect to BIDC. In individual kinetic runs, plots of log[BIDC] versus time were linear (r2 > 0.996). Further, the pseudo-first- order rate constants do not depend on the initial concentration of BIDC (Table 2). The order with respect to furfural was less than one (Table 2). A plot of k
obs
versus [furfural] is shown in Figure 1. The downward curvature of the plots suggests the existence of a complex. A plot of 1/k
obs
versus 1/[furfural] was linear with an intercept on the rate ordinate (Figure 2). Thus, a Michaelis–Menten type kinetics was observed with respect to furfural. This leads to the postulation of following overall mechanism and rate law: A plot of 104 kobs versus [furfural]. [BIDC] = 0.0005 mol L-1; [H+] = 1.0 mol L-1; temperature 308 K A plot of 1/(104kobs) versus 1/ [furfural]. [BIDC] = 0.0005 mol L-1; [H+] = 1.0 mol L-1; temperature 308 K.





Formation constants and thermodynamic parameters of furfural-BIDC complexes.
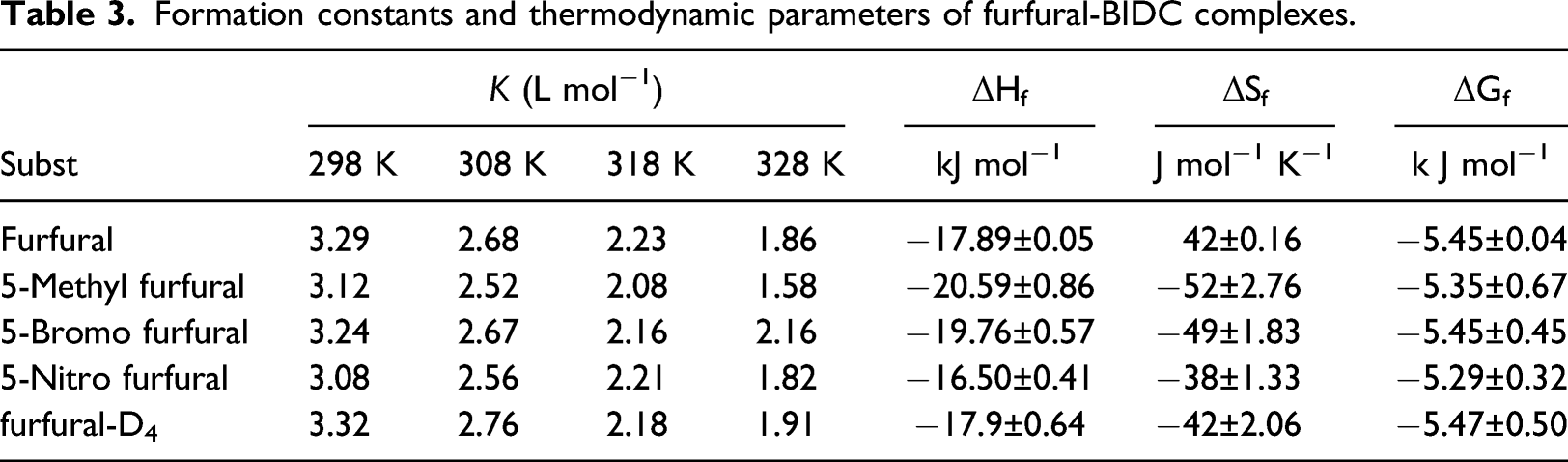
Rate constants of decomposition of furfural-BIDC complexes and their activation parameters.

Spectral analysis
Inspection of the spectra of BIDC and BIDC+furfural showed that there is a definite increase in the absorbance of BIDC on addition of furfural (Figure 3). UV-VIS spectra of [A] 0.0005 mol L-1 BIDC, [B] 0.0005 mol L-1 BIDC + 0.001 furfural, [C] 0.0005mol L-1 BIDC + 0.015 furfural, [D] 0.0005 mol L-1 BIDC + 0.025 furfural. Solvent: DMSO, temperature: 298 + 1 K.
Kinetic isotope effect
To establish the importance of the cleavage of α-C−H bond in the rate-controlling step, the oxidation of deuterated furfural (furfural-D4) was studied with BIDC. The findings (Tables 3 and 4) showed that the formation constants of the complexes of normal and deuterated furfural are nearly close but the rates of their decomposition indicated a primary isotope effect (kH/kD = 6.23 at 298 K). In the oxidation of benzaldehyde by BIDC 12 , nearly similar value of primary kinetic isotope effect of ca. Six was obtained. Further, in the present study, the value of kinetic isotope effect decreases with an increase in temperature.
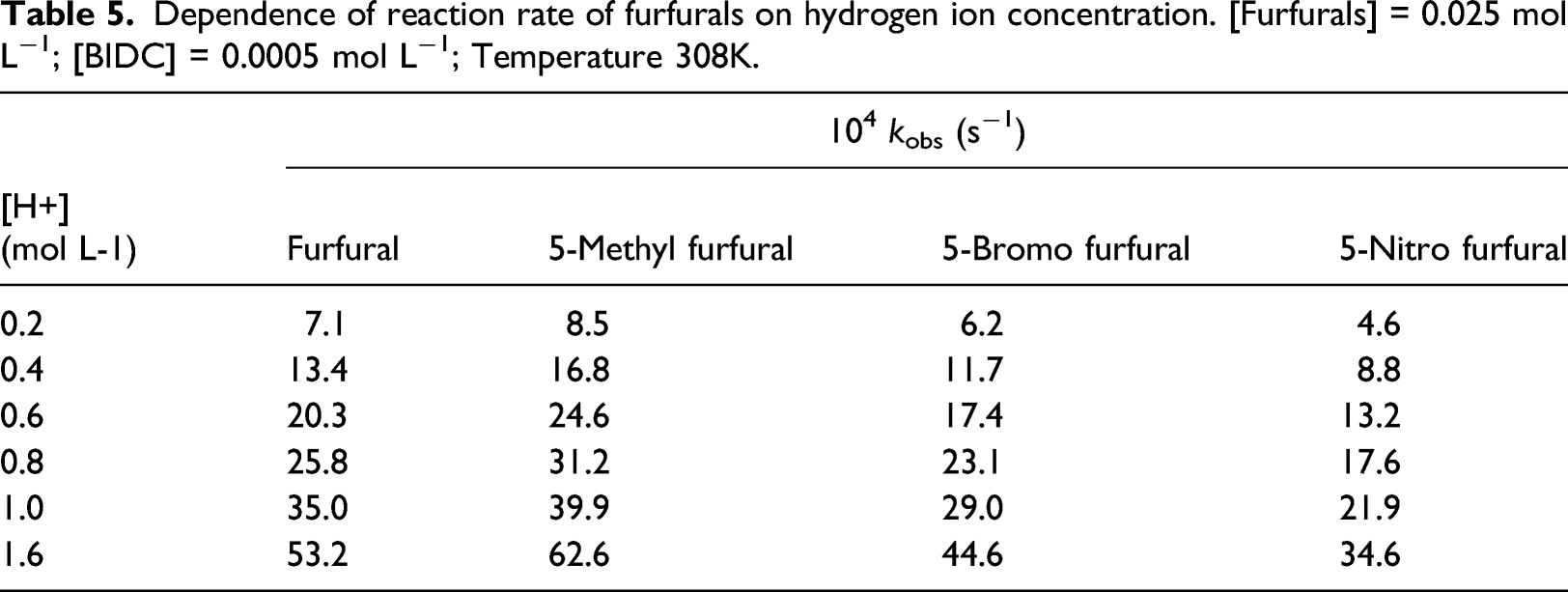
Effect of acidity
The rates of oxidation favoured with an increase in acidity and the dependence is of the form: Rate = k´ [H+] (Table 4). For all furfurals, order with respect to [H+] is one (Figure 4). A plot of log k
obs
versus log[H+], for all furfurals, exhibited the slope >0.96 and r2 > 0.99. A plot of 104k
obs
versus [H+] at 308 K. [BIDC] = 0.0005 mol L−1 [furfural] = 0.025 mol L−1; temperature 308 K.
Dependence of reaction rate of furfurals on hydrogen ion concentration. [Furfurals] = 0.025 mol L−1; [BIDC] = 0.0005 mol L−1; Temperature 308K.
Effect of substituents
The kinetics of oxidation of some 5-substituted furfurals were studied in DMSO medium in presence of TsOH. The effect of substituent on rate is explained on the basis of resonating structures of furfural.
In these structures fifth carbon atom of furfural is an electron-deficient center. Therefore, the rate of oxidation is accelerated by electron donating substituent and retarded by electron withdrawing substituent at fifth carbon of furfural. The order of reactivity is as follows: 5-methyl > 5-Hydrogen > 5-Bromo > 5- Nitro
Solvent effect
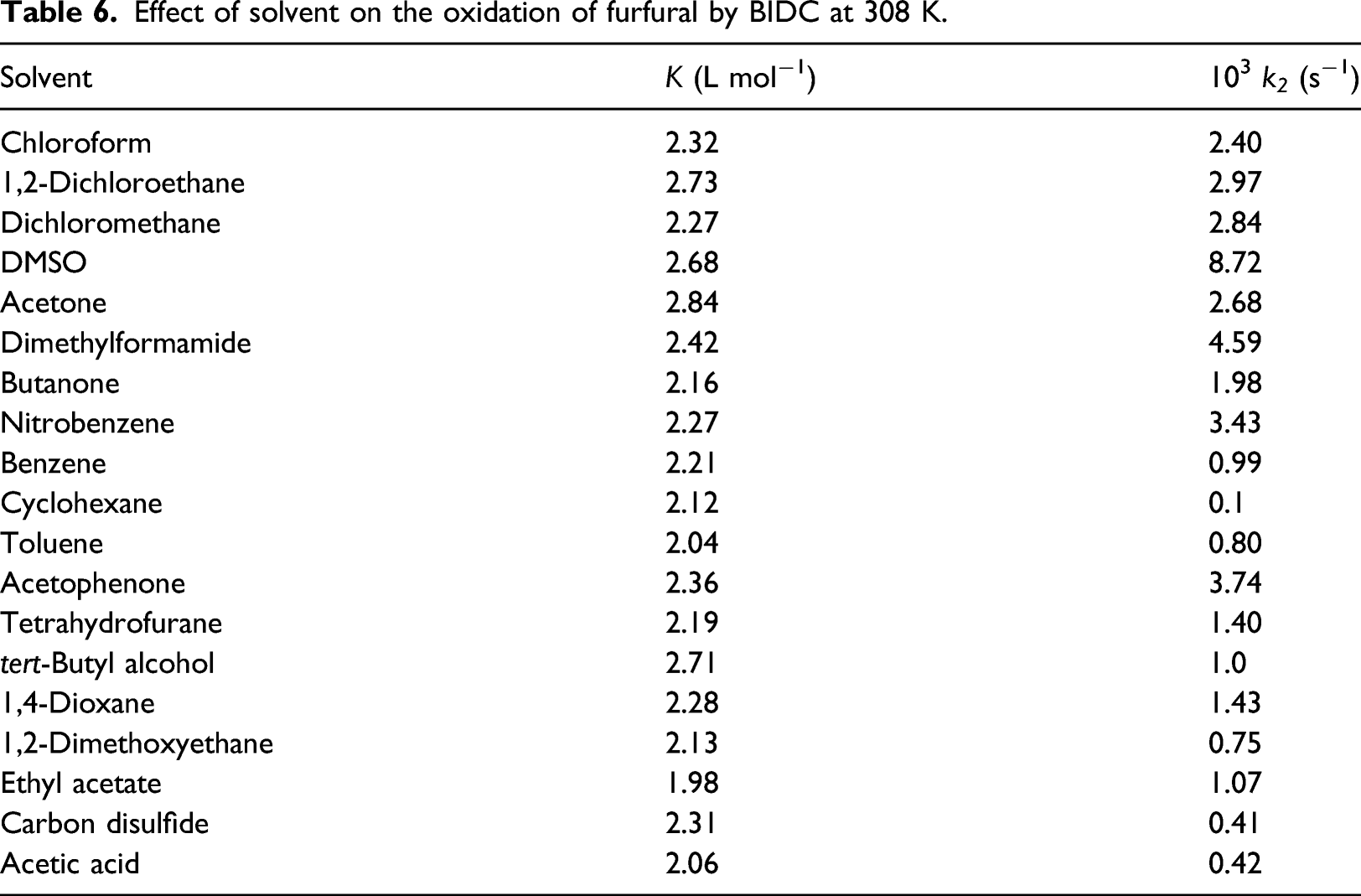
Effect of solvent on the oxidation of furfural by BIDC at 308 K.
Discussion
Conductivity measurements have been carried out at 308 K to determine the state of BIDC in our reaction conditions. It was noticed that DMSO has very low conductivity and the inclusion of BIDC in DMSO shows insignificant variation in the conductivity value. Therefore, under our reaction conditions, BIDC can be considered as non-ionised and does not break up as dichromate and benzimidazolium ions. The rate of oxidation does not alter on addition of benzimidazolium ion, also favours the postulation that BIDC remains as non-ionised 13 . 28 reported the crystal structure study of BIDC, supports the non-ionic character of the oxidant in the reaction system. Two benzimidazolium rings attached with the dichromate ion via. Hydrogen bonds. BIDC forms a number of hydrogen bonds with the hydrogen donor (N-H) and hydrogen acceptor (O) in the molecule. Further, an intermolecular hydrogen bridge is unusually formed between two adjacent dichromate ions. By these hydrogen bridges the molecules are combined into infinite chains which controlled the releasing procedure of dichromate ions to the reaction system and hence the compound acts as non-ionic in our reaction system.
Solvent effect
The data described in Table 6 represents that the equilibrium constant, K # , is quite insensitive with change in solvent, however, k2, varies considerably. Hence, the rate constant of the decomposition of complexes, k2, in 18 solvents (CS2 was not studied as the absolute span of the solvent parameters are unavailable), were correlated in terms of linear solvation energy relationship (LSER) of 29 . But the correlations were irrelevant.
The solvent effect data were studied in connection with Swain’s
30
equation (6), where ampere denotes the anion-solvating power of the solvent and B denotes the cation-solvating power and solvent-polarity is denoted by the term (A + B) where intercept term is denoted by C.
The conclusion of the correlation analysis in respect of Swain’s equation (6), individually with ampere and B, and with (A + B) are described as

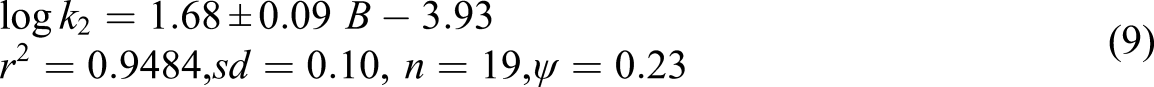

The observed results of solvent effect revealed a perfect correlation regarding Swain’s equation 30 with both cation- and anion-solvating powers participating to the observed solvent effect. Although, the contribution of cation-solvation is greater, it alone contributes for ca. 95% of the data (A + B), represents the solvent polarity, explains ca. 82% of the data. Relative permittivities of the solvents were correlated with the data by considering the fact that ca. 82% of the data is contributed by (A+B). A plot of log k 2 versus 1/D, where D represents relative permittivity of the solvent, though, It is not linear (r2 = 0.5068). This showed that the relative permittivity and solvent polarity, defined by 30 do not represent the same solvent characteristics.
Mechanism
Considering the absence of any effect of acrylonitrile on the rate of reaction, it is unfavoured that free radical oxidation is operative in the present reaction
31
. BHT is a perfect trapping reagent for free radicals. However, BHT was recovered unchanged also goes in opposition to the existence of free radical oxidation. The formation constants of the complexes of normal and deuterated furfural are nearly same but the rates of their decomposition showed a primary kinetic isotope effect. This specifies that in furfural, the aldehydic C–H bond is cleaved in rate controlling step. Formation of electron-deficient reaction centre in the transition state is favoured by analysis of the solvent effect indicating major contribution of the cation-solvation on the rate of decomposition of the complex. It is supported by reactivity order of furfurals. Therefore, it is indicated that the removal of hydrogen as hydride-ion results in an electron-deficient species in rate controlling step. However, the observed Michaelis - Menten type kinetics with respect to furfural led to propose the formation of 1:1 complex by a nucleophilic attack of aldehydic oxygen on chromium in a fast pre-equilibrium (Scheme 1). The nature of complex suggested is similar to that reported in oxidation of aromatic aldehyde by BIDC
12
. The observed order of reactivity indicated that the electron-donating group at five position of furan ring, speed up the oxidation process. This is accounted in terms of an increase in electron-availability at oxygen of the aldehydic group resulting in the facilitation of the complex formation. The observed acid-dependence of the reaction points to a fast reversible protonation of the intermediate complex proceeding to its disproportionation. A mechanism represented in account for the experimental results. Based on the proposed mechanism, rate law can be written as- Mechanism of oxidation of furfural by BIDC.

With the help of Equations (4) and (11), we have, k 2 = ka K1 [H+].
Ampere hydride-ion transfer may occur either by a chromate ester or via an acyclic process. Kwart and Nickle [32] developed a method to analysis of the temperature dependence of the kinetic isotope effect, showed that the loss of hydrogen proceeds via a concerted cyclic mechanism. The data for ordinary and deuterated-furfural were expressed in a familiar manner
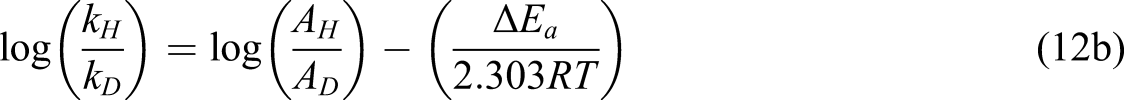
The activation energy difference for kH/kD is 4.59 kJ mol−1 which concluded by final results agrees with the zero-point energy gap for the C-H and C-D bonds (ca. 4.53 kJ mol−1) and the entropy of activation (ΔS*) of the respective reactions are almost same. This directly corresponds to the properties of a symmetrical transition state33,34. Earlier, in the oxidation of benzaldehydes by BIDC 12 , similar kinds of results have been noticed. Bordwell 35 has given strong evidence against the phenomenon of concerted one-step bimolecular process of hydrogen shifting. It is strongly recommended in the present reaction system that the hydrogen transfer does not take place by an acyclic bimolecular process. The truly symmetrical processes involving linear transfer of hydrogen are intrinsically concerted sigmatropic reactions characterized by transfer with a cyclic transition state 36 . The shifting of two electrons in a cyclic system of the reaction was the next step of the reaction. This electrocyclic mechanism for the oxidation of furfural by BIDC involved six electrons, being a Hückel type (4n+2)π electron system, is an allowed process 37 . Hence, one can conclude that in the oxidation of furfural by BIDC, the hydride-ion shifting occurs via a cyclic transition state. The manner of electron transfer has to be set up. The first step includes the nucleophilic attack of aldehydic-oxygen electrons on electron-deficient chromium atom to form an intermediate complex. This complex goes.
Through unimolecular decomposition in the slow step. The transition state involves the bonding of hydrogen atom to both the OH group attached to chromium and the aldehydic-carbon. The electron flow in a cyclic transition state has been observed assuming that hydrogen is separated as hydride-ion. Therefore, the way of electron transfer occurs through the carbon-hydrogen-oxygen-chromium bond (Scheme 1). This would develope the initiation of carbcationic species by reverting back the nucleophilic attack of aldehydic oxygen. Similar kind of mechanism has noticed in the oxidation of aliphatic aldehydes by BIDC 11 .
The proposed mechanism was favoured by the observed loss of entropy of activation. The two ends become highly solvated when the charge separation occurs in the transition state. In the transition state, this results in an immobilization of an immense number of solvent molecules, exhibited in the loss of entropy. The negative entropy also accounts for the effect of solvent.
Initially oxidation state of Chromium metal reduced from Cr(VI) to Cr(IV) and expected to react with another Cr(VI) to produced Cr(V) which is ultimately reduced in a fast step to produce Cr(III). Such a pattern of reactions in Cr(VI) oxidations is well established 38 .
It is interesting here to compare the findings of the oxidation of the existing reaction with the results of oxidation of furfurals by other Cr(VI) complexes. The oxidation of furfural by IDC 15 , QCC 17 and QFC 18 in 60% acetic acid–water medium, exhibited first order with substrate and oxidant. However, order with respect to hydrogen ion concentration is two. The rate of oxidation increases with increase in percentage of acetic acid. The retardation of rate by addition of Mn+2 ions conforms that a two electron transfer process is involved in the reaction. The kinetics of the oxidation of furfural by BBI 14 , QDC 16 and INDC 20 in 50% (v/v) aqueous acetic acid leading to 2-furoic acid. The reaction was found to be first order with respect to each of oxidant, furfural and hydrogen ion. The oxidant was converted into the protonated dimetallic Cr(VI) species. The kinetic data are explained with reference to the aldehyde-hydration equilibrium. The observed results support a mechanistic pathway proceeding via a rate-controlling oxidative decomposition of the chromate ester of the aldehyde hydrate. However, Sekar et al. reported oxidation by chromic acid [18] in 70% (v/v) aqueous acetic acid exhibited first order with respect to each of oxidant, furfural and acid. Similar kind of kinetics was observed with NDC 19 in 30% (v/v) aqueous acetic acid. The comparative study of oxidation kinetics of furfural by NBN and NCN 22 in acetic acid medium exhibited first order with respect to substrate and inverse first order with respect to oxidant and hydrochloric acid. The reaction slowed down on adding one of the reaction products, nicotinamide (NA). The rate of oxidation increases with acetic acid percentage. Thus, it is noticed that the oxidation process depends on the nature of the oxidant and the reactions conditions.
Conclusions
The oxidation of furfural by BIDC in DMSO resulted in the formation of 2-furoic acid. The site of oxidation reaction was carbonyl function. The reaction exhibited first order kinetics with respect to hydrogen ion and oxidant. Michaelis–Menten type kinetics observed with furfural. A α-C-H bond breaking is indicated in rate controlling step. A mechanism involving the rate-controlling oxidative decomposition of complex through a hydride-ion transfer from furfural to oxidant to give end product is suggested.
Footnotes
Acknowledgements
We thank University Grants Commission, New Delhi, India, for their financial support. Further, sincere thanks to Prof. Seema Kothari, Department of Chemistry, J.N.V. University, Jodhpur, for her valuable suggestions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.