
Abstract
Reactions of aryl 1-(2,4-dinitronaphthyl) ethers with piperidine in dimethyl sulfoxide at 25oC resulted in substitution of the aryloxy group at the ipso carbon atom. The reaction was measured spectrophotochemically and the kinetic studies suggested that the titled reaction is accurately third order. The mechanism is began by fast nucleophilic attack of piperidine on C1 to form zwitterion intermediate
Introduction
The reactions of piperidine with aromatic nitro compounds containing poor leaving group in dimethyl sulfoxide (DMSO) have been reported to subject base catalysis.1–5 The uncatalyzed pathway proceeded by slow formation of zwitterion followed by fast step to the protonated product which equilibrated with the substitution product (Scheme 1). The catalysis has been reported to be either the slow proton transfer step, 6 that is, specific base (SB)2,7–13 or the rate-limiting step is the removing of the leaving group by assistance of the conjugate acid,9,14–18 SB-GA (Scheme 1). The change in rate controlling step has been reported to be dependent on the base strength and the basicity of the leaving group.9,19 The differentiation between the two pathways mechanism of catalysis is achieved using external base related to SB catalysis and conjugate acid of the same amine used in the reaction related to SB-GA catalysis.
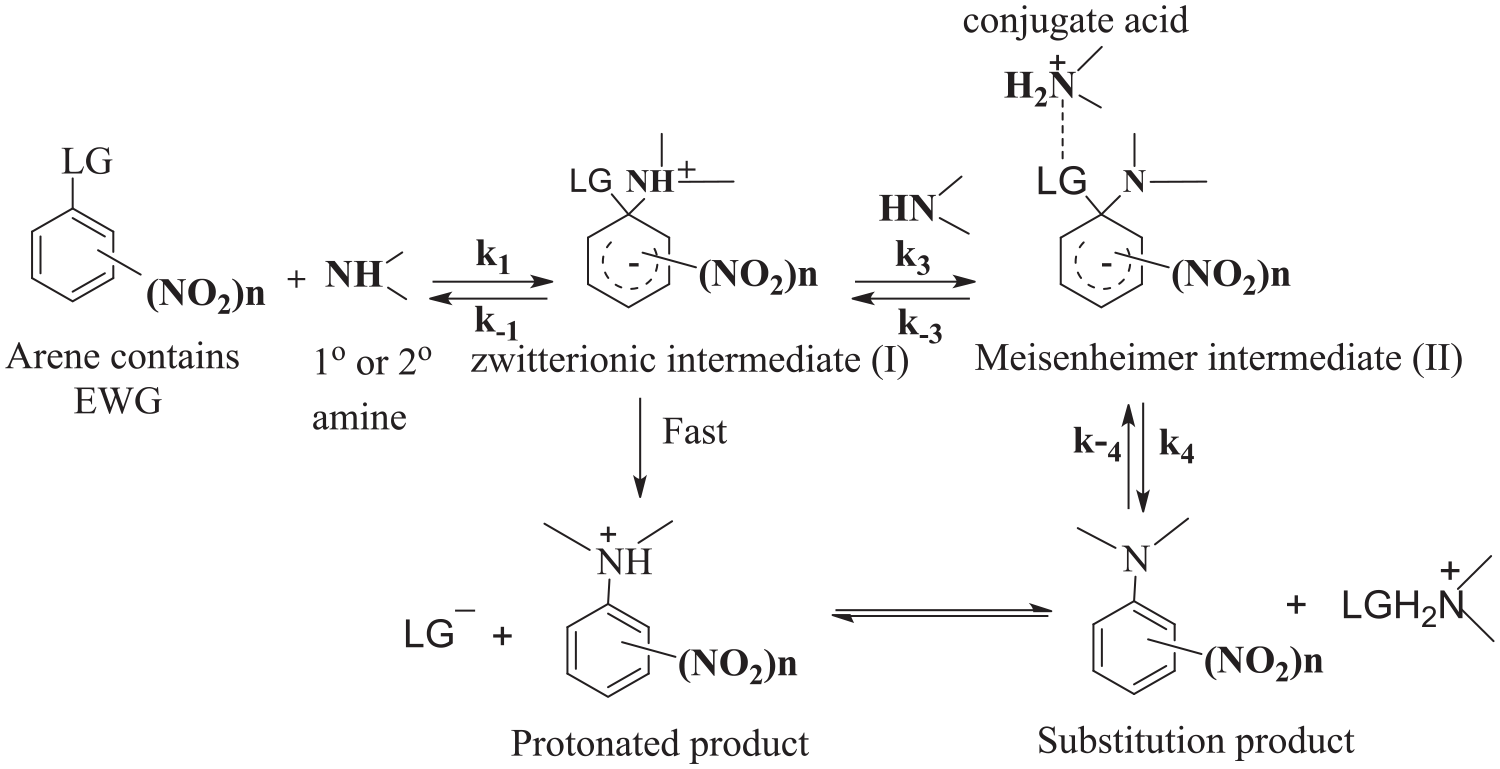
Base catalyzed and uncatalyzed Mechanism of aminolysis of aromatic nitro compounds containing leaving group.
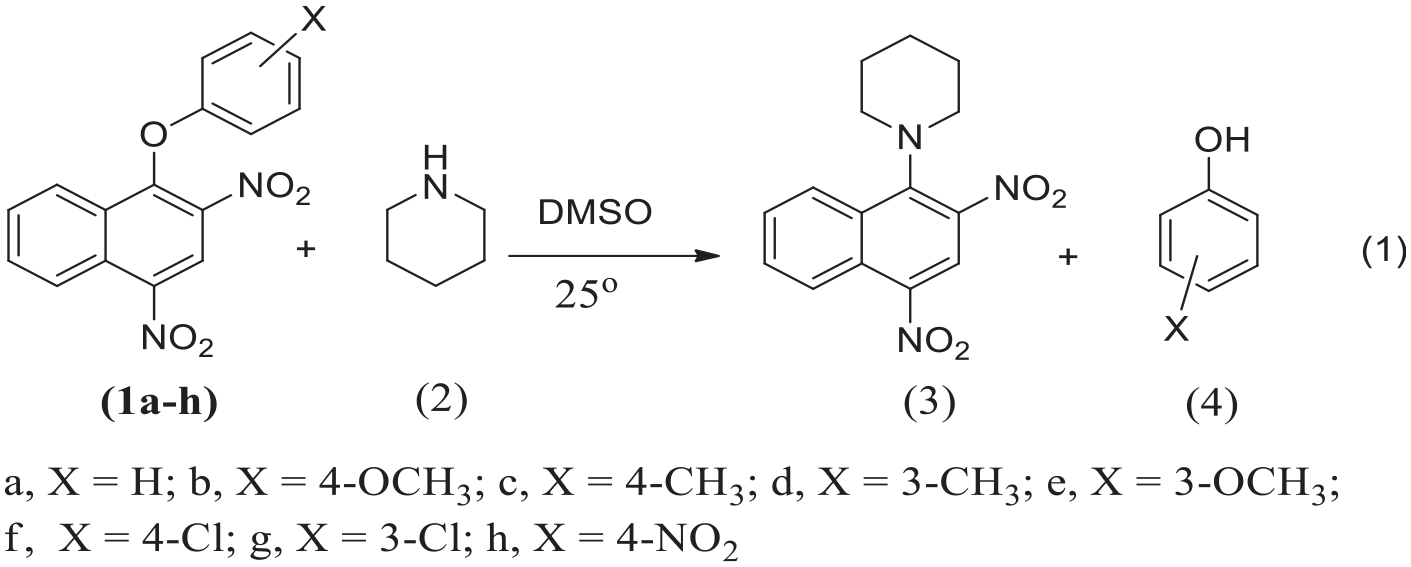
Several studies have been reported that aryl 1-(2,4-dinitronaphthyl) ether
Experimental
Preparation of aryl 1-(2,4-dinitronaphthalen-1-yl) piperidine (3)
The reaction 1-aryl 2,4-dinitro-1-naphthyl ethers
Kinetic technique
Spectrophotometric studies
The reaction of 1-aryl 2,4-dinitronaphthyl ether
Kinetic measurements
A solution of
Method of calculations
All computational calculations had been performed on personal computer using the Gaussian 09W program packages and 6.31G(d,p) basis set
36
Gaussian output files were visualized by means of Gaussian view 05 software.
37
Computation provided useful information about the optimized molecular structures of
Discussion
Structure determination of 1-(N-piperidinyl)-2,4-dinitronaphthalene, 3
The reaction between ethers
Optimized geometry of piperidine, (2)
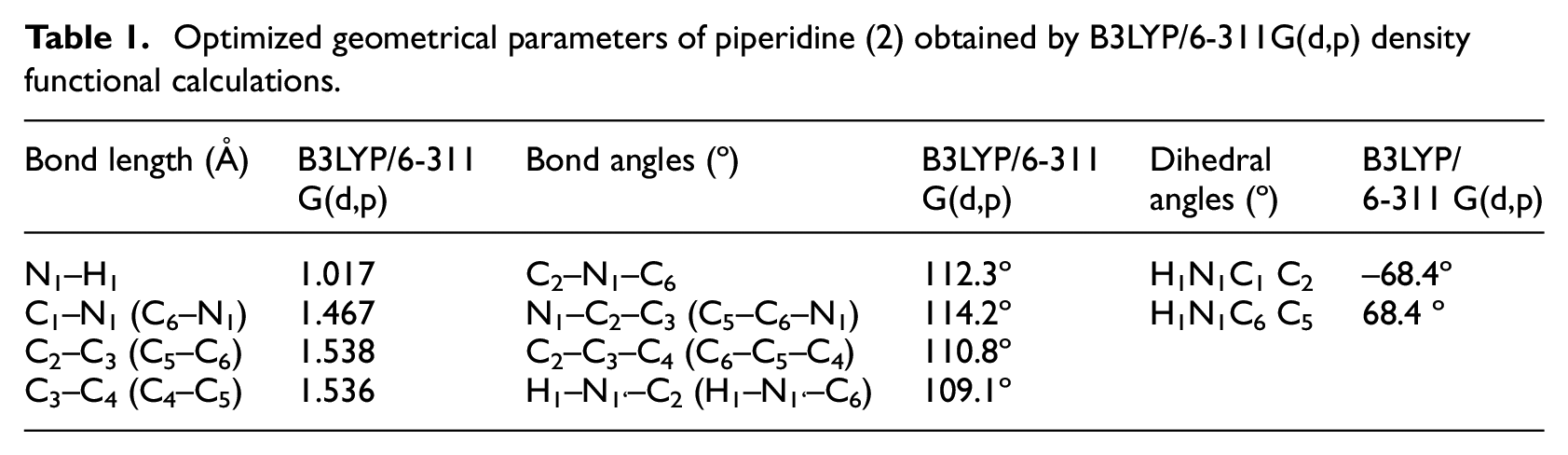
Piperidine molecule is a heterocyclic amine and has two possible chair conformations.38,39 The calculated and experimental vibrational modes of piperidine had been reported.40–47 The optimized geometric parameters (bond lengths and angles) by BLYP with 6-311G(d,p) are in accordance with the atom numbering given in Figure 1 and Table 1.
Optimized geometry and numbering of piperidine (2).
Optimized geometrical parameters of piperidine (2) obtained by B3LYP/6-311G(d,p) density functional calculations.
Molecular orbital analysis of piperidine (2) and (1a–h)
The density functional theory (DFT)
48
was used to understand the chemical reactivity and site selectivity of 1-(2,4-dinitronaphthyl) ether
Mulliken charge, NBO charge, and atomic orbital coefficient of HOMO in piperidine (2).
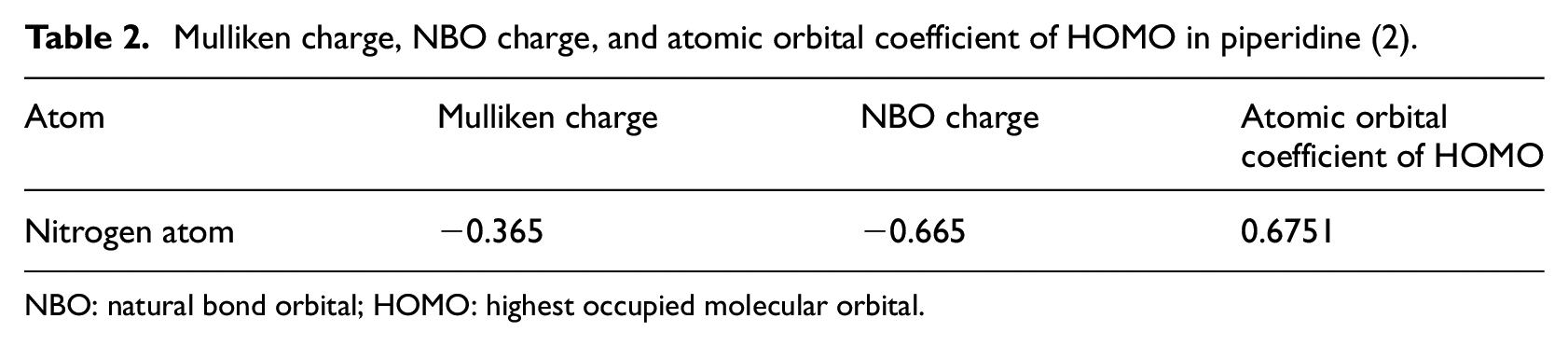
NBO: natural bond orbital; HOMO: highest occupied molecular orbital.
The global and local chemical reactivity descriptors49,50 were calculated from HOMO and lowest unoccupied molecular orbital (LUMO) energies of
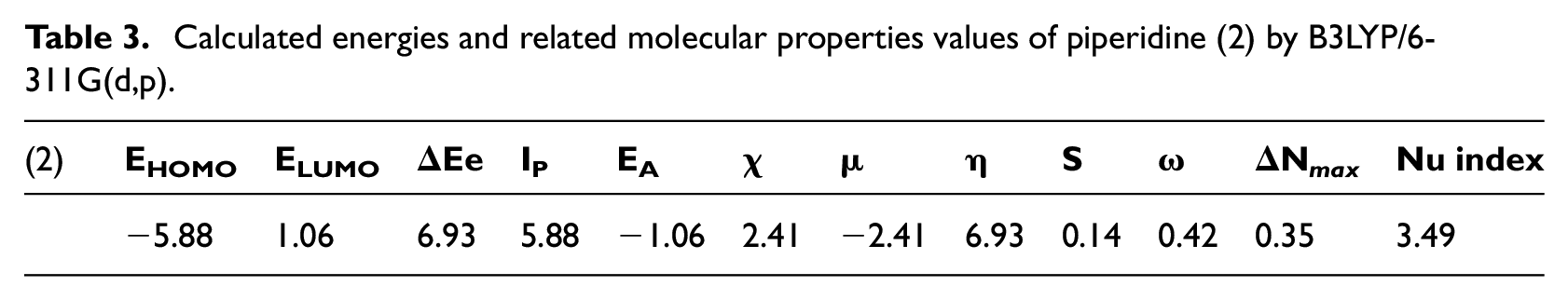
Calculated energies and related molecular properties values of piperidine (2) by B3LYP/6-311G(d,p).
Calculated energies and related molecular values of aryl 2,4-dinitronaphthyl ether (1a–h) by B3LYP/6-311G(d,p).
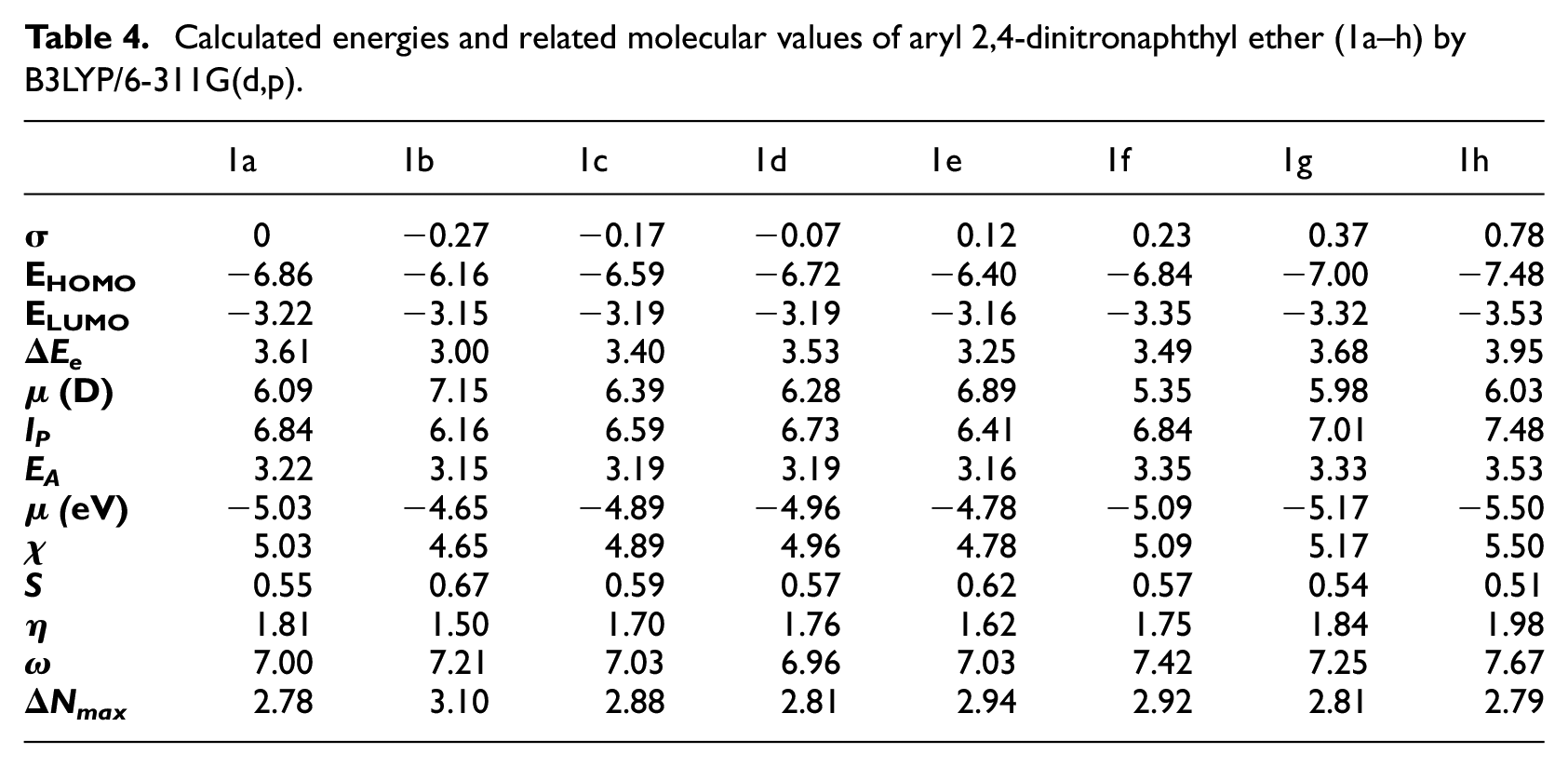
Table 3 gives the energy of HOMO, energy of LUMO, chemical potential, hardness, softness, electrophilicity index and nucleophilicity index of piperidine (
The calculated energies and related molecular properties values of
Mulliken, NBO atomic charge, and atomic orbital coefficient of LUMO for the selected centers (C1, C1’) calculated by B3LYP/6–311G(d,p) for aryl 2,4-dinitronaphthalene ethers (
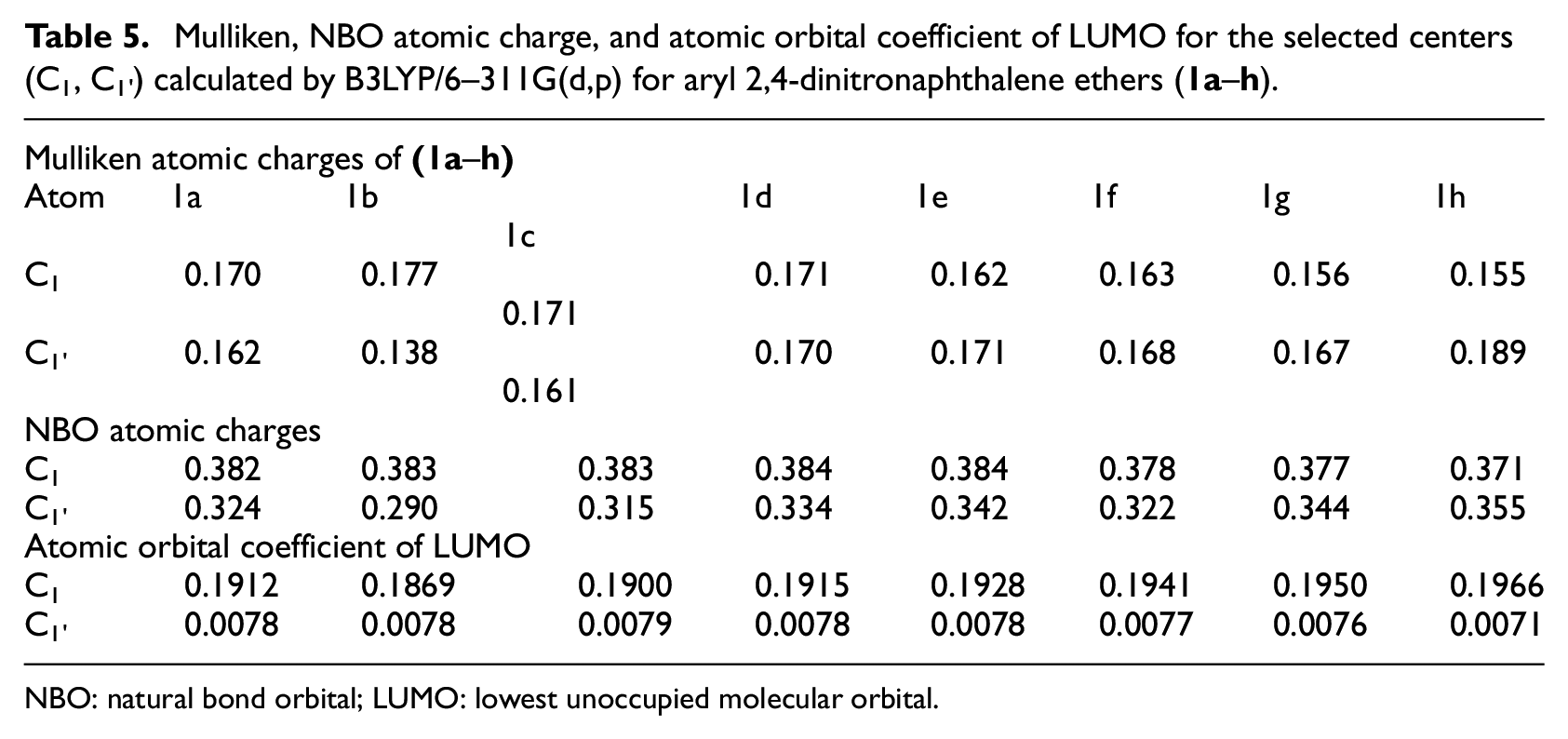
NBO: natural bond orbital; LUMO: lowest unoccupied molecular orbital.
The atomic charge values and atomic orbital coefficients are important in determining the reactivity of reaction centers toward nucleophilic attack.
62
Table 5 showed that (1) the naphthyl ipso carbon C1 was more positively charged than the aryl ipso carbon C
The mechanism for the piperidinolysis of ethers
Difference between the two possible HOMO/LUMO combinations for (1a–h) and piperidine (2).

HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital.
The energy gap between the HOMO and LUMO is very important in determining the chemical reactivity of the molecule. The high value of the energy gap indicates that the molecule shows high chemical stability, while a small HOMO–LUMO gap means small excitation energies to the manifold of excited states. The reported global parameters of ethers (
These parameters confirm that
The local nucleophilic attack f
Local reactivity descriptors
FF 49 is one of the widely used local density functional descriptors to model chemical reactivity and site selectivity. The condensed FF of piperidine was calculated using the procedure proposed by Yang and Mortier 64 based on a finite difference method (equations (2)–(4)).
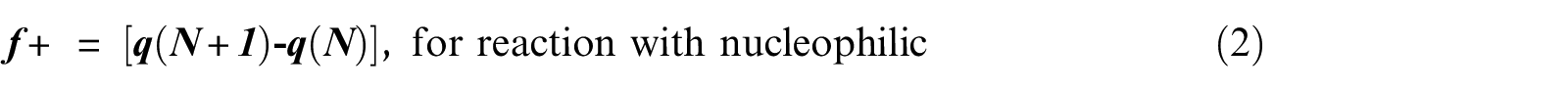

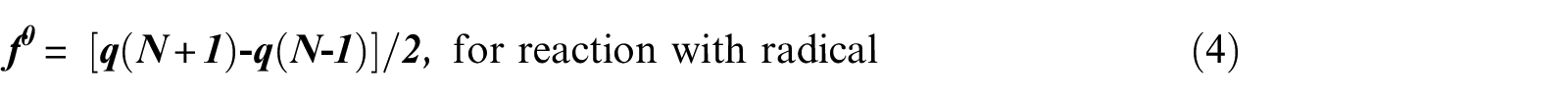
where


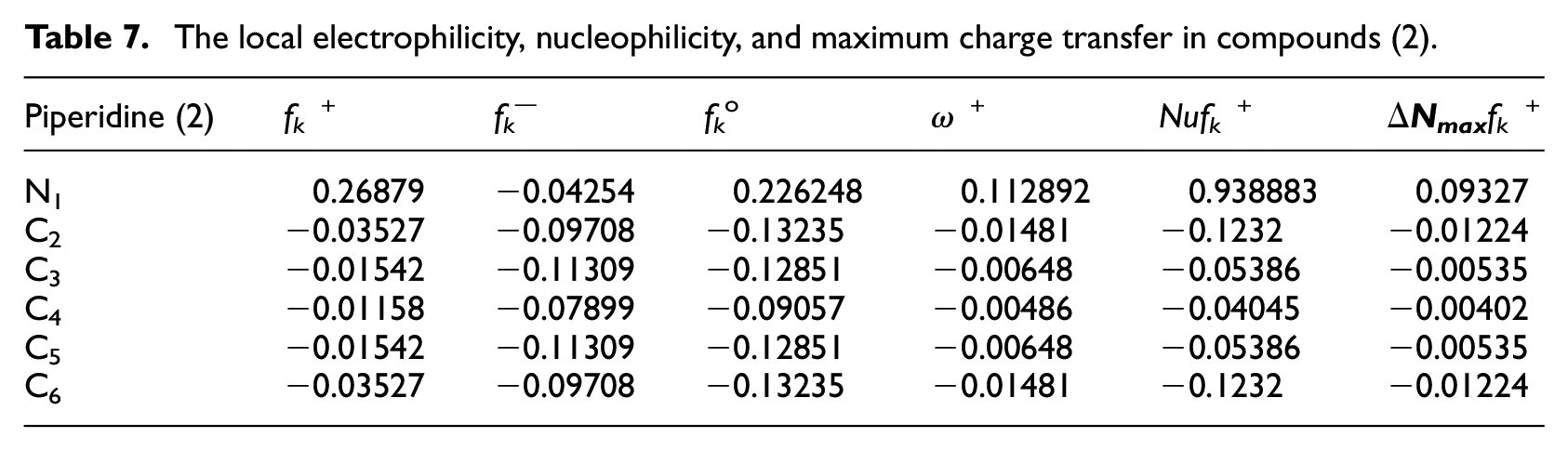
The local electrophilicity, nucleophilicity, and maximum charge transfer in compounds (2).
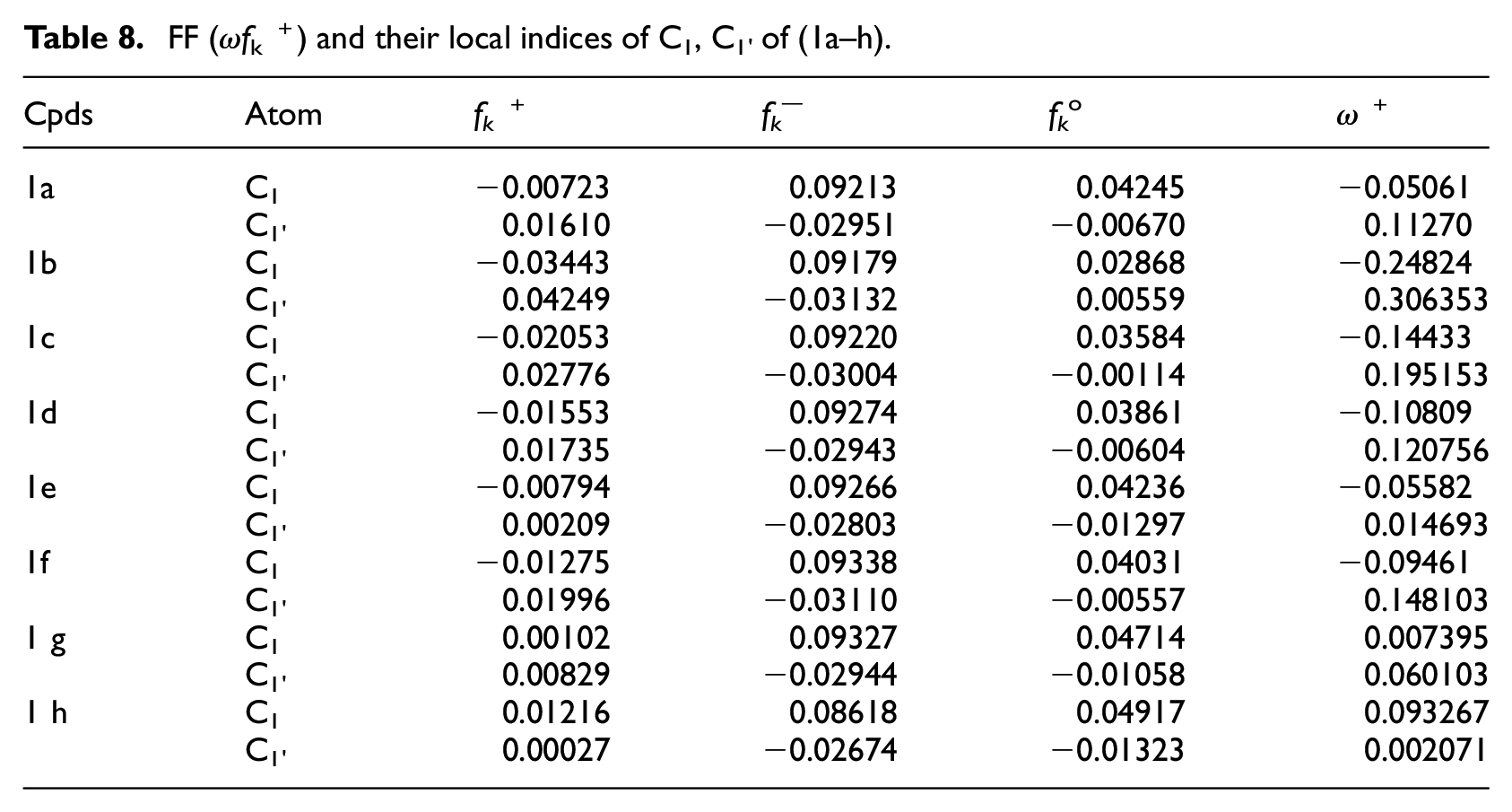
FF (ωfk +) and their local indices of C1, C1’ of (1a–h).
The maximum charge transfer can be written as follows57,59

The
Experimental kinetic studies for the reaction of aryl 1-(2,4-dinitronaphthyl)ether (1a–h) with piperidine (2) in DMSO at 25oC
The kinetic studies for the reaction of aryl 1-(2,4-dinitronaphthyl) ether
Search for piperidine catalysis. Pseudo first-order (kobs) and third-order rate constants (kN) for the reaction of compounds (1 × 10-4 M/L) (1a–h) with piperidine (2) DMSO at 25oC.
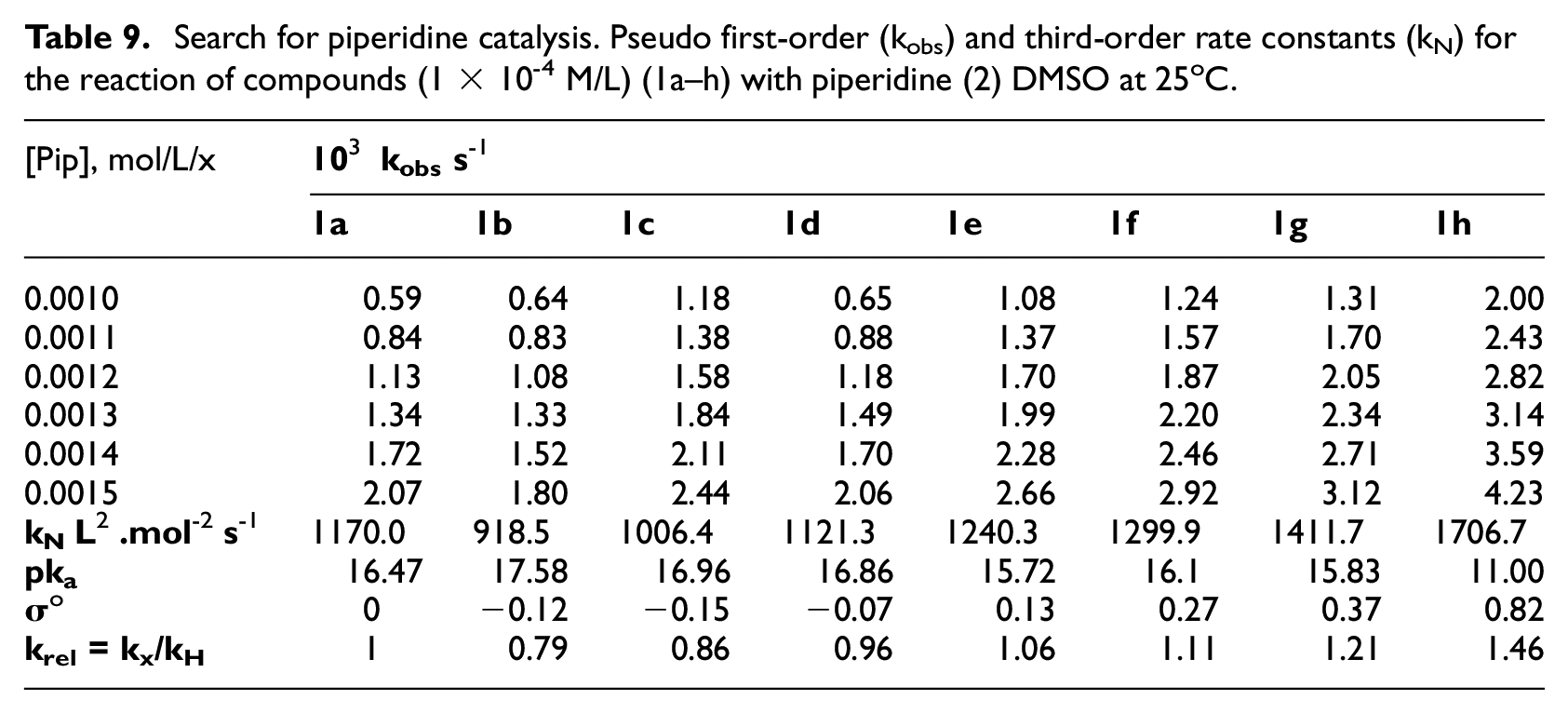
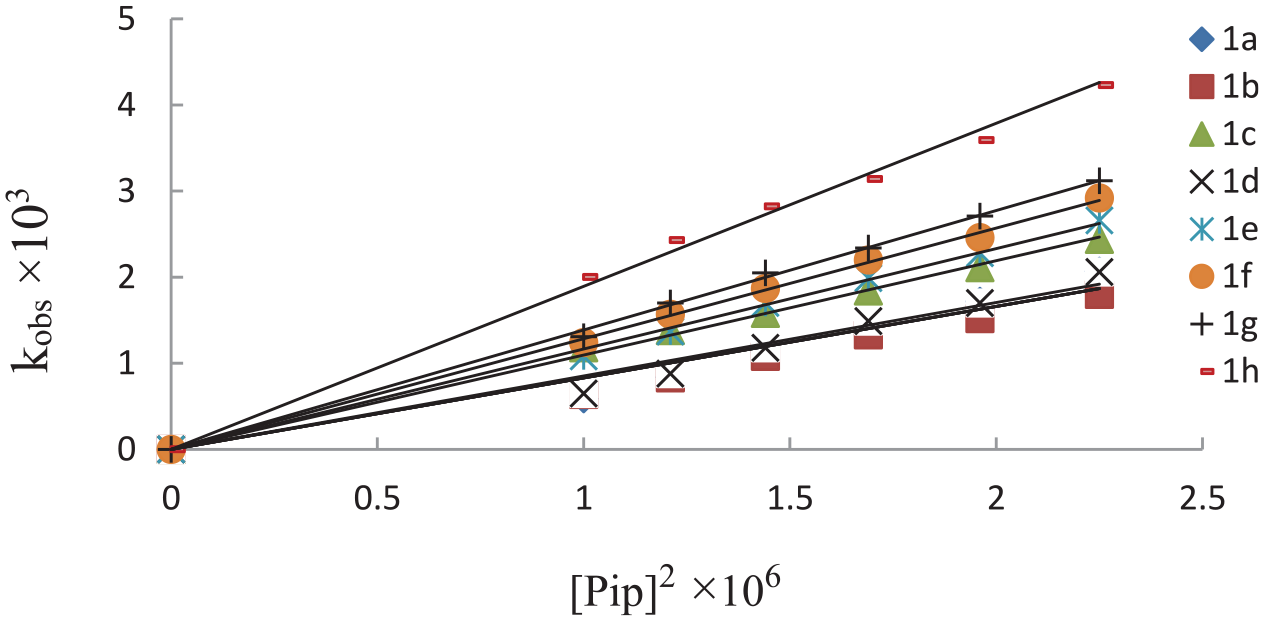
Plots showing linear dependence of kobs on [piperidine] 2 for the reaction of aryl 1-(2,4-dinitronaphthyl) ether (1a–h) with piperidine (2) at 25oC.
The third-order rate constants (kN) at 25oC for the formation of the piperidino product
The assignment of the piperidino product (
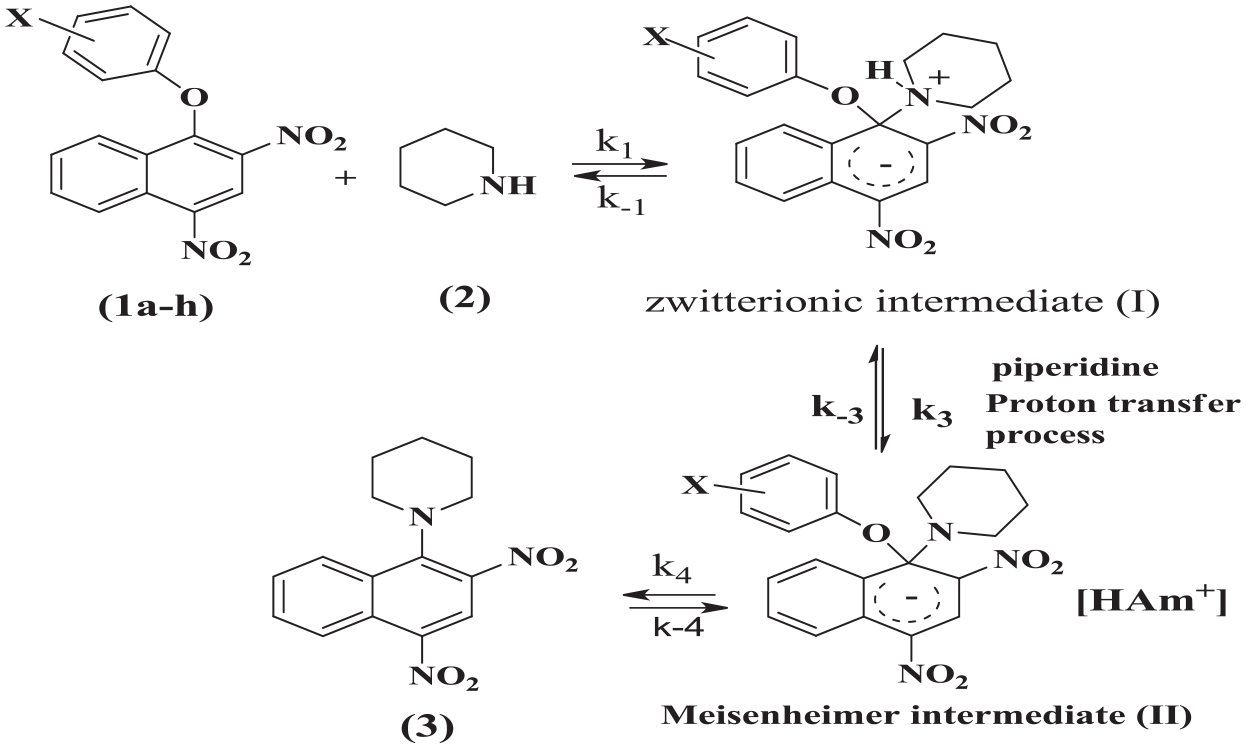
Reaction of aryl 1-(2,4-dinitronaphthyl) ethers (1a–h) with piperidine (2).
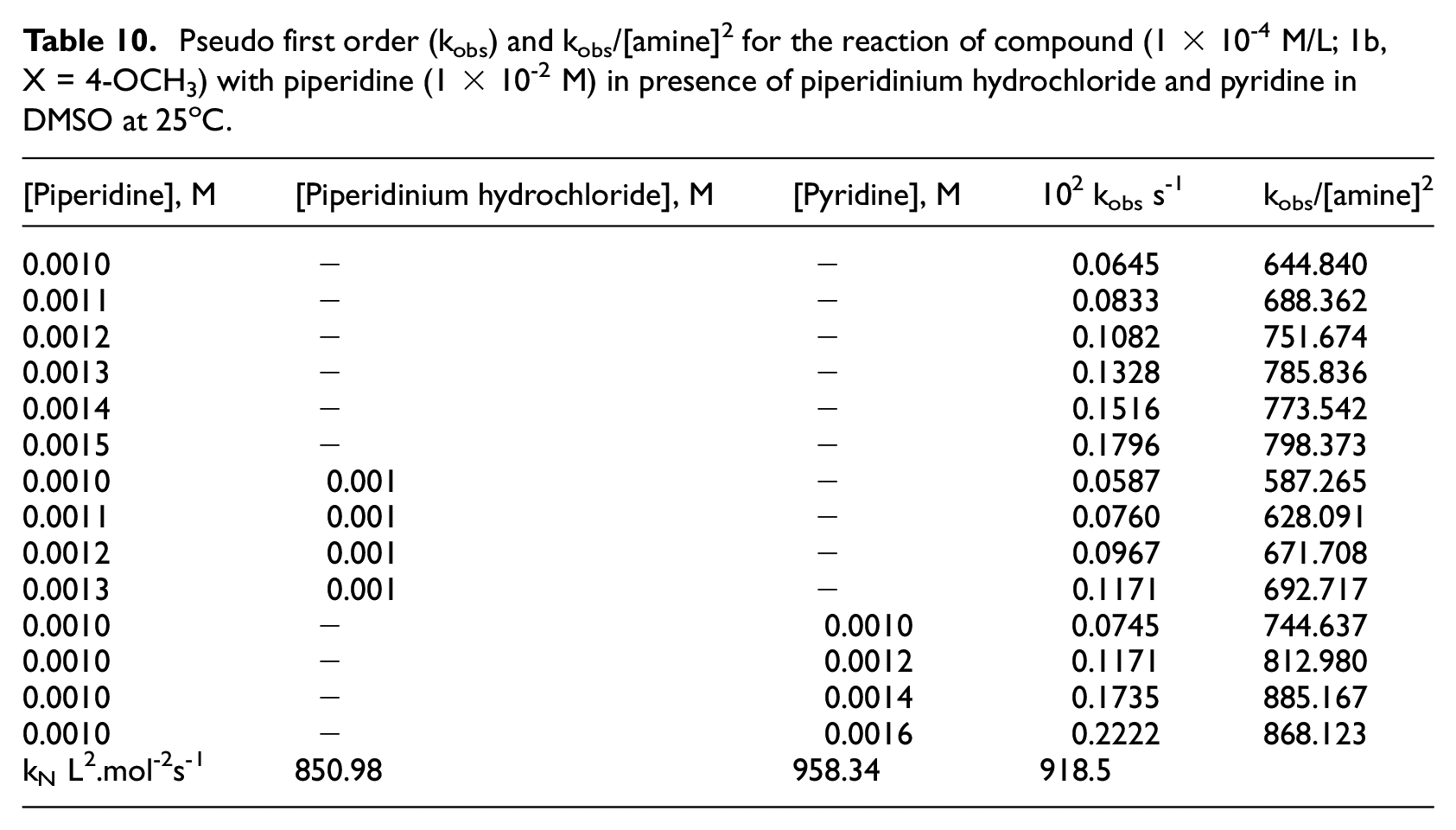
The small krel differences, shown in Table 9, in addition to the product analysis and order of the reaction proposed that the proton transfer step were slow. This suggestion was ensured by study the effect of addition external base, such as pyridine, or in the presence of conjugate acid, such as piperidinium hydrochloride. The catalysis is pronounced by the addition of external base pyridine, while the presence of conjugate piperidinium ion showed negligible change in rate constants for reaction of
Pseudo first order (kobs) and kobs/[amine] 2 for the reaction of compound (1 × 10-4 M/L; 1b, X = 4-OCH3) with piperidine (1 × 10-2 M) in presence of piperidinium hydrochloride and pyridine in DMSO at 25oC.
Rate equation for the reaction of ethers (1a–h) with piperidine (2) to form 1-piperidino-2,4-dinitronaphthalene (3)
The overall rate equation (equation (8)) was derived according to
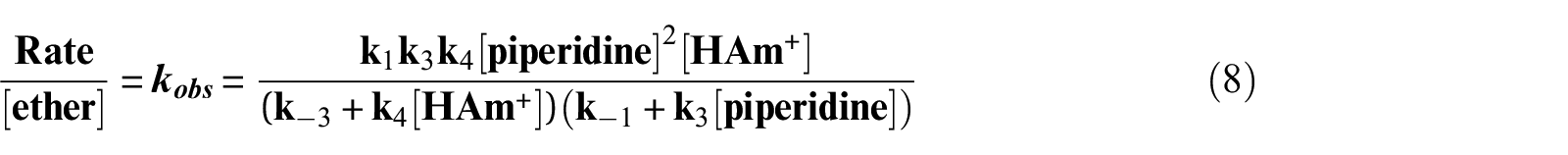
Since the third-order values was increased by addition of external base, therefore, the removing of the phenoxy (the k4 step) was fast and equation (8) is reduced to equation (9)

where k3 is the third-order rate constant. Thus, proton transfer was rate determining step, k-1 >> kAm[Am] and kN equal k1k3/k-1.
Structure–reactivity relationships
The application of Hammett concept
The rate constants were correlated with different σo-Taft’s constant values for substituted phenyl ring. 68 The σo-Taft’s values represented inductive constants for substituted phenyl groups (–Ar) relative to the unsubstituted one C6H5–. Therefore, the electronic effect of the substituents in the leaving group moiety could be quantified by the use of a Hammett equation (10), 69 σo is the substituent constant and ρ is the reaction constant.

Plot of log kN versus σo-Taft’s 68 gave good straight line with ρ value of + 0.247 with correlation coefficients (r = 0.91). The linearity of Hammett plot was a good evidence for the same mechanism for all substituents in the titled reactions, while ‘ρ’ of + 0.247 pointed out a poor electronic effect of the substituent. 70
The small positive value of ρ may be attributed to the compensation between the opposite charges in the activated state of the slow step between zwitterion (

And, the Hammett equation could be written in the form of equation (12).

The small positive value of ρ showed that the activated state of the slow step was resembled to the product (Meisenheimer intermediate). It has been reported that the high
The application of Brønsted concept
The magnitude of the Brønsted coefficient has usually been related to the extent of bond formation in the activated complex that involved in the slow step (equation (13)). 72

The magnitude and the sign of Brønsted coefficient (β) was reported to depend on the pKa either for attacking nucleophile or the leaving group.73,74 As the pKa of the nucleophile varied with constant leaving group a positive Brønsted coefficient (βN) is observed.24,32 On the other hand, the change of the pKa values of the leaving group with constant nucleophile would result in a negative Brønsted coefficient (βlg) 30 due to the inverse proportionality between pKa of the leaving group and the rate.
Table 7 showed that the reactivity of
Computational studies of mechanism for the reaction of pheny1 1-(-2,4-dinitronaphthyl) ether (1a) with piperidine (2)
The identification of transition states and their existence were confirmed by the presence of a single imaginary frequency in the Hessian matrix.77–79 The kinetic results proved that the substitution reaction was overall third order and catalyzed by the second molecule of piperidine, that is, SB-GA (Scheme 2). Therefore, the mechanism pathway was rewritten to show the fine processes (
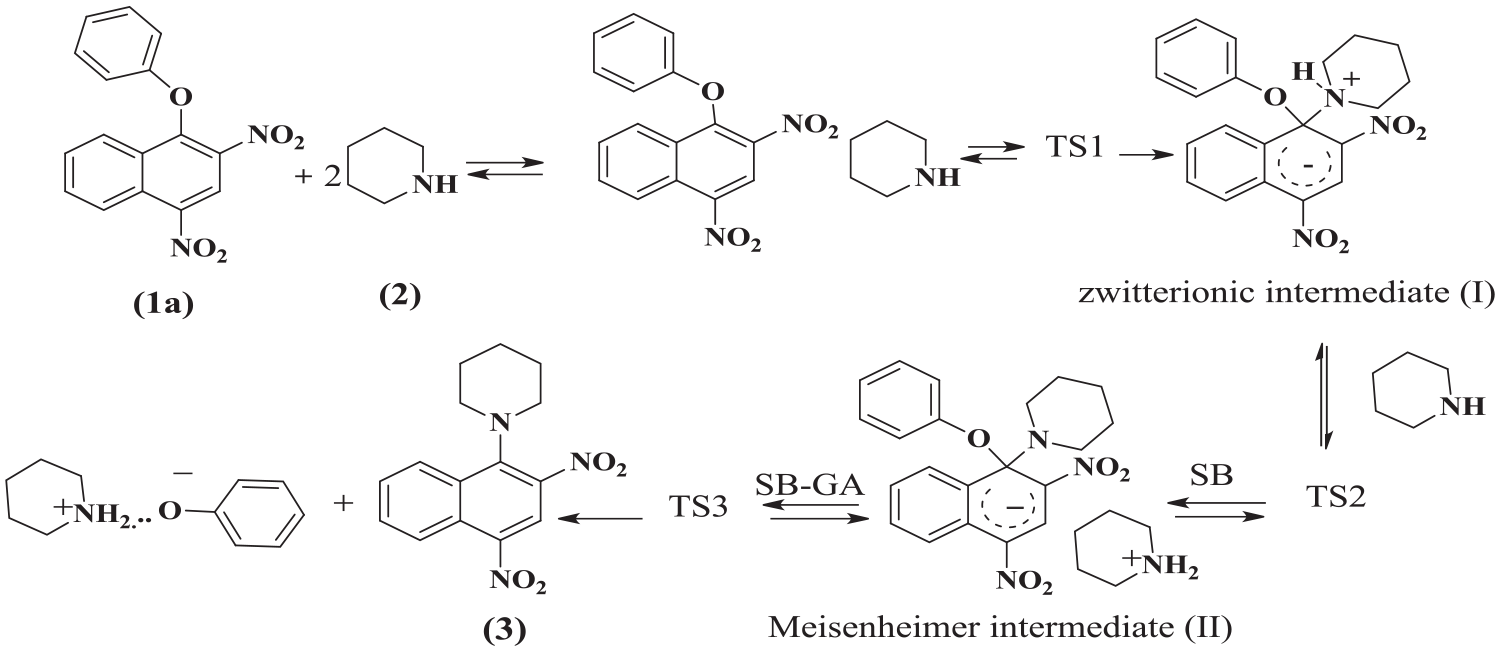
The mechanism of ether (1a) with piperidine (2).
Energy profile and geometrical analyses of activated complexes involved in the piperidinolysis of (1a–h)
The theoretical mechanism of catalyzed reaction, the energies of the reactants, the products, and the activated states were calculated by DFT methods were determined (Figure 3). It expressed the possible activated complexes for the reaction of (
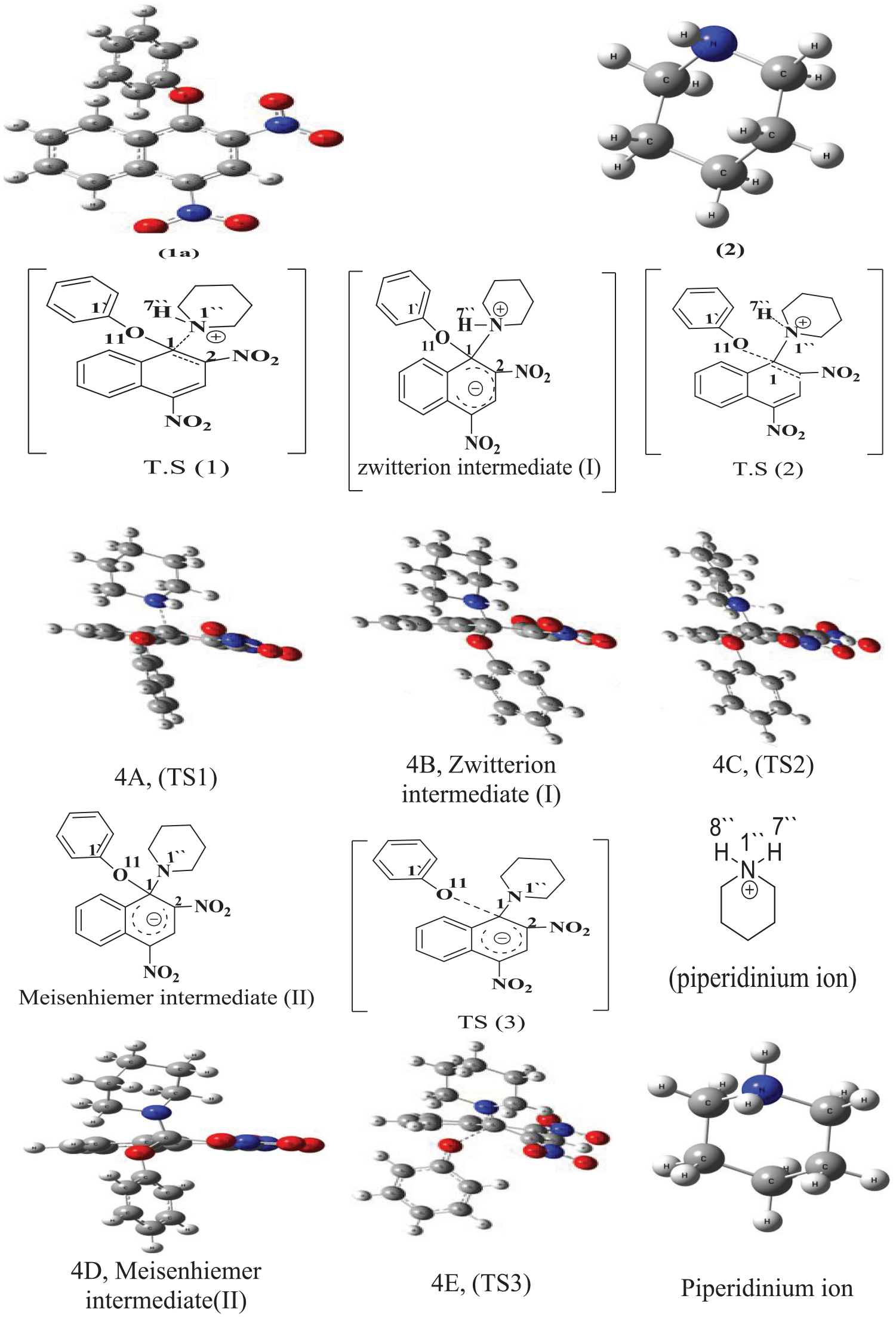
B3LYP/6-311G(d,p) optimized geometries of the transition states and intermediates involved in the piperidinolysis of (1a–h) in DMSO.
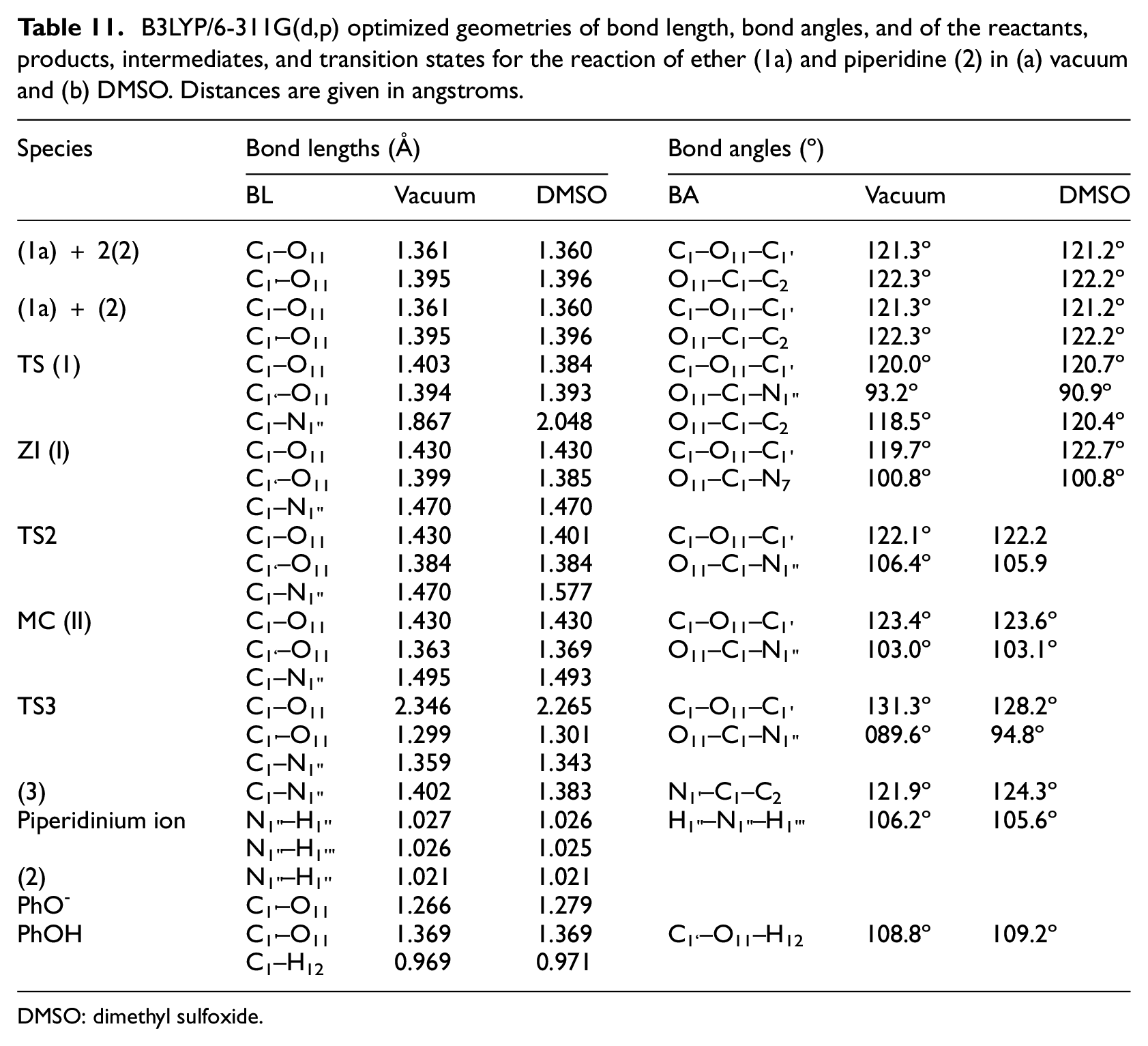
B3LYP/6-311G(d,p) optimized geometries of bond length, bond angles, and of the reactants, products, intermediates, and transition states for the reaction of ether (1a) and piperidine (2) in (a) vacuum and (b) DMSO. Distances are given in angstroms.
DMSO: dimethyl sulfoxide.
The bond length of C1–O11 of zwitterion intermediate (
Pictures of the ether (1a), phenol, and phenoxide anion in DMSO.
The bond lengths of C1–O11 and C1–N1’’ in TS2 had the same bond lengths as those in zwitterion intermediate
Energy values of the possible activated complexes shown in Figure 4 were depicted in Table 12. The activation energies of all species and compounds in the reaction of
Energy (in hartree/particle) of the reactants, products, intermediates, and transition states for the reaction of ether (1a) and piperidine (2) in (a) vacuum and (b) DMSO.

DMSO: dimethyl sulfoxide.
Conclusion
The reaction of aryl 1-(2,4-dinitronaphthyl) ether (
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.