
Abstract
The reaction mechanism between silacyclopropenylidene and three-membered heterocyclic compounds (azirane and oxirane) has been systematically investigated at the B3LYP/6-311+G* level of theory in order to better understand the reactivity of unsaturated cyclic silylene. Geometry optimizations and vibrational analyses have been conducted for the stationary points on the potential energy surface of the system. Calculations show that the Si-spiroheterocyclic intermediate and four-membered heterocyclic silylene compound could be produced through the insertion process and subsequent dissociation process between silacyclopropenylidene and three-membered heterocyclic compounds. For the insertion process, it is easier for silacyclopropenylidene to insert into C-N bond of azirane than into C-O bond of oxirane. This study is helpful to understand the reactivity of silacyclopropenylidene, the evolution of silicon-bearing molecules in space, and to offer an alternative approach to the formation of enlarged heterocyclic silylene compound.
Introduction
Silicon is one of the 10 most abundant elements in the cosmos and hence silicon-bearing molecules are important in astrochemistry.1–3 Eleven silicon-bearing molecules have been detected in space corresponding to almost 10% of the total number of known astronomical molecules. 4 Since the silicon-bearing molecules played a vital role in the chemical evolution of the interstellar medium and the formation of silicon carbide dust grains in the circumstellar envelopes of carbon-rich stars like IRC+10216, 5 the molecular processes involved in the formation of organosilicon molecules such as silicon carbide (SiC) and silicon dicarbide (SiC2) together with their hydrogenated counterparts (C2H2Si) have received considerable attentions from the astronomical and physical chemistry communities in recent years.6–9 Owing to the similarity of C2H2Si to know astronomical molecules (SiC, SiC2), those of composition of C2H2Si might be abundant in space as well. 10
As an analogue of C3H211,12 and C2NH, 13 C2H2Si is an example of highly unsaturated silylene. Preliminary experimental evidence for C2H2Si isomers in the laboratory was reported by Maier’s group.14,15 By means of the pulsed flash pyrolysis of the gaseous mixture of 2-ethynyl-1,1,1-trimethyldisilane and argon, silacyclopropenylidene (c-C2H2Si) was isolated in an argon matrix at 10 K. As displayed in Scheme 1, irradiation of matrix-isolated silacyclopropenylidene resulted in a series of photochemical rearrangements leading to ethynylsilanediyl (HCCSiH), vinylidenesilanediyl (H2CCSi). Based on theoretical calculations, silacyclopropenylidene is the most stable C2H2Si species.16,17
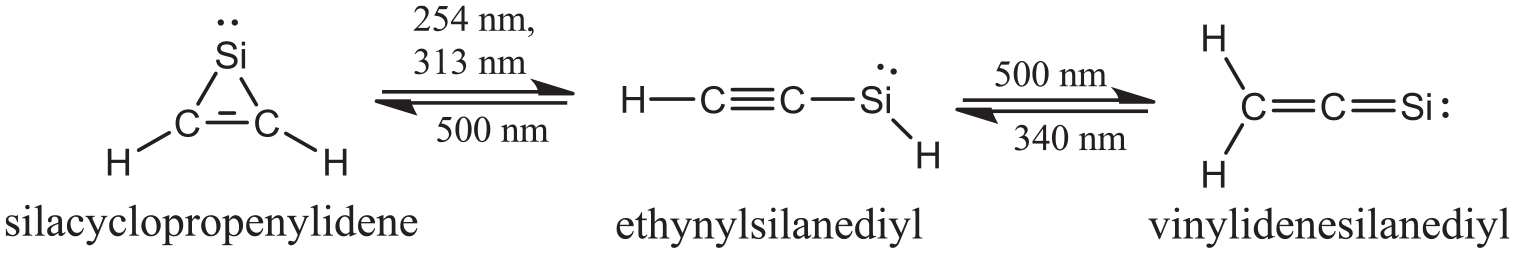
The transformation of three C2H2Si isomers.
There are numerous theoretical and experimental investigations on the C2H2Si isomers.18–20 In addition, the relative stability, structures, and dipole moments of the C2H2Si isomers have been investigated extensively. 21 Wu et al. 22 performed an anharmonic vibrational analysis of the electronic ground states of the three C2H2Si isomers employing second-order vibrational perturbation (VPT2) theory. The relative stability and structural characteristics of silyenic C2HXSi 23 and dialkyl substituted C2X2Si 24 species have been calculated at different levels of theory. The microwave rotational spectrum of vinylidenesilanediyl and its isotopic species were observed in a pulsed supersonic molecular beam by Fourier transform microwave spectroscopy. 25
The bimolecular collision of the simplest silicon-bearing radical silylidyne (SiH) with acetylene (C2 H2) and ethylene (C2 H4) can be considered as the prototype mechanism leading to the formation of organosilicon molecules in circumstellar envelopes.26,27 These reactions could supply the molecular feedstock of organosilicon molecules necessary to account for the ubiquitous presence of interstellar silicon-carbide grains via a “bottom up” synthesis starting with small silicon-bearing precursor molecules as proposed here. However, the reactions of the most stable C2H2Si species, silacyclopropenylidene, have not been reported up to now. In the present study, we have performed a comprehensive theoretical investigation of the reaction mechanism between silacyclopropenylidene and three-membered heterocyclic compounds (azirane and oxirane) by employing density functional theory (DFT) method in order to better understand the silacyclopropenylidene reactivity. For the reaction partner, oxirane has been previously detected in the interstellar medium, especially in star-forming regions.28,29 However, unlike oxirane, azirane has not been detected in space up to now although both of them have similar geometric features. This study is helpful to understand the reactivity of silacyclopropenylidene, the evolution of silicon-bearing molecules in space, and to offer an alternative approach to the formation of enlarged heterocyclic silylene compound.
Calculation methods
The popular hybrid density functional B3LYP method, namely Becke’s three-parameter non-local exchange functional 30 with the non-local correlation functional of Lee et al., 31 and 6-311+G* basis set including diffuse and polarization functions have been employed to locate all the stationary points along the reaction pathway without imposing any symmetry constraints. The reliability and efficiency of this method in predicting the geometries and properties have been verified by a number of investigations.32–34 Frequency analyses have been conducted to confirm the nature of the minima and transition states. Moreover, intrinsic reaction coordinate (IRC) calculations have also been made to further validate the calculated transition states connecting reactants and products. In addition, the relevant energy quantities, such as the reaction energies and barrier heights, have been corrected with zero-point vibrational energy (ZPVE) corrections.
To further refine the calculated energy parameters, single point energy calculations for the reaction have been performed at the CCSD(T)/6-311+G* level of theory based on the optimized geometries at the B3LYP/6-311+G* level of theory. As outlined in Table 1, two levels can give consistent results for the calculated reaction profile. For the sake of simplicity, the energetic results at the B3LYP/6-311+G* level of theory have been mainly discussed below if not noted otherwise.
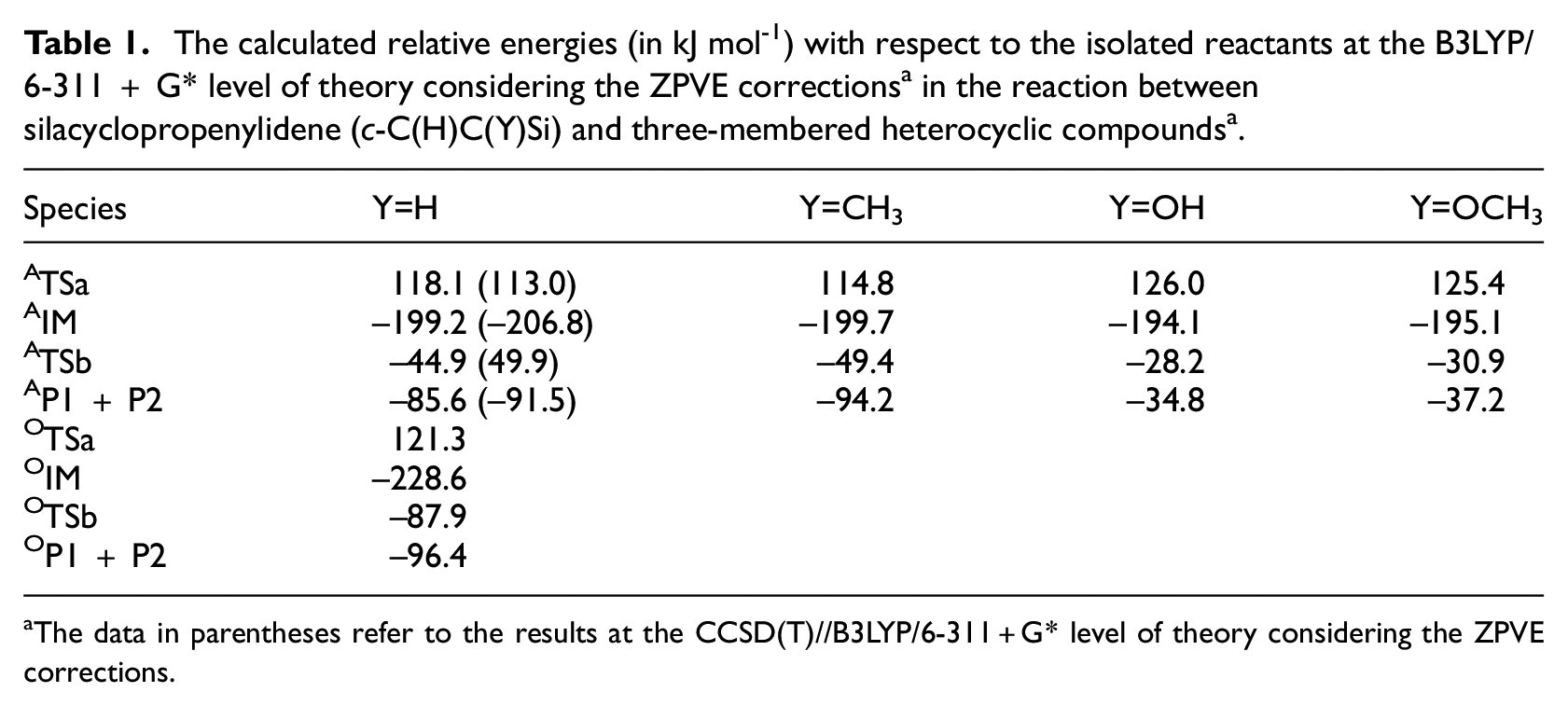
The data in parentheses refer to the results at the CCSD(T)//B3LYP/6-311+G* level of theory considering the ZPVE corrections.
All the calculations have been performed using Gaussian 98 program. 35
Results and discussion
Considering the structural similarity between azirane and oxirane, we will mainly discuss the reaction mechanisms based on the reaction between silacyclopropenylidene and azirane, which are marked with superscript “A.” As for the reaction of silacyclopropenylidene and oxirane, we marked it with superscript “O.”
As displayed in Scheme 2, the probable pathway for the title reaction has been proposed. The geometric parameters for the reactants (R1-silacyclopropenylidene, R2-azirane or oxirane), transition states (TS), intermediate (IM), and products (P1-azasilacyclobutylidene or oxasilacyclobutylidene, P2-acetylene) involved in the pathway are displayed in Figure 1. The calculated relative energies for the available stationary points and the corresponding reaction profile are illustrated in Figure 2.
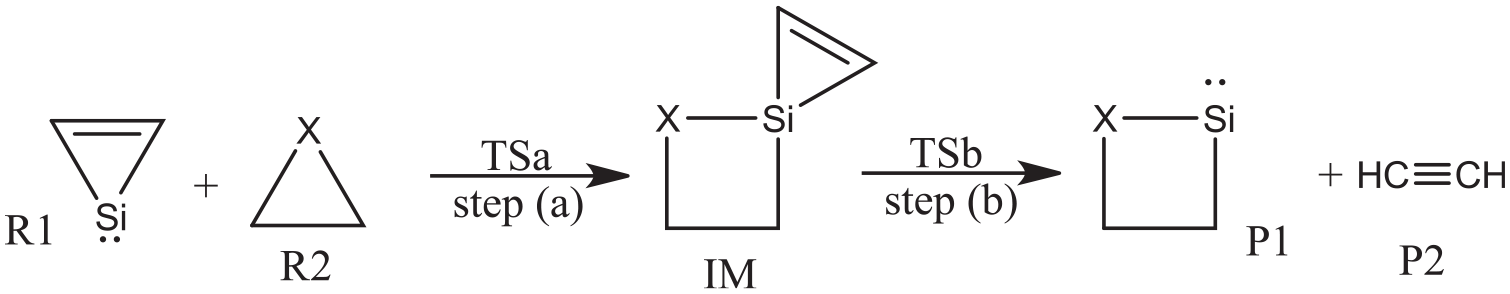
The proposed pathway for the reaction between silacyclopropenylidene and three-membered heterocyclic compounds c-CH2CH2X (X=NH, O).
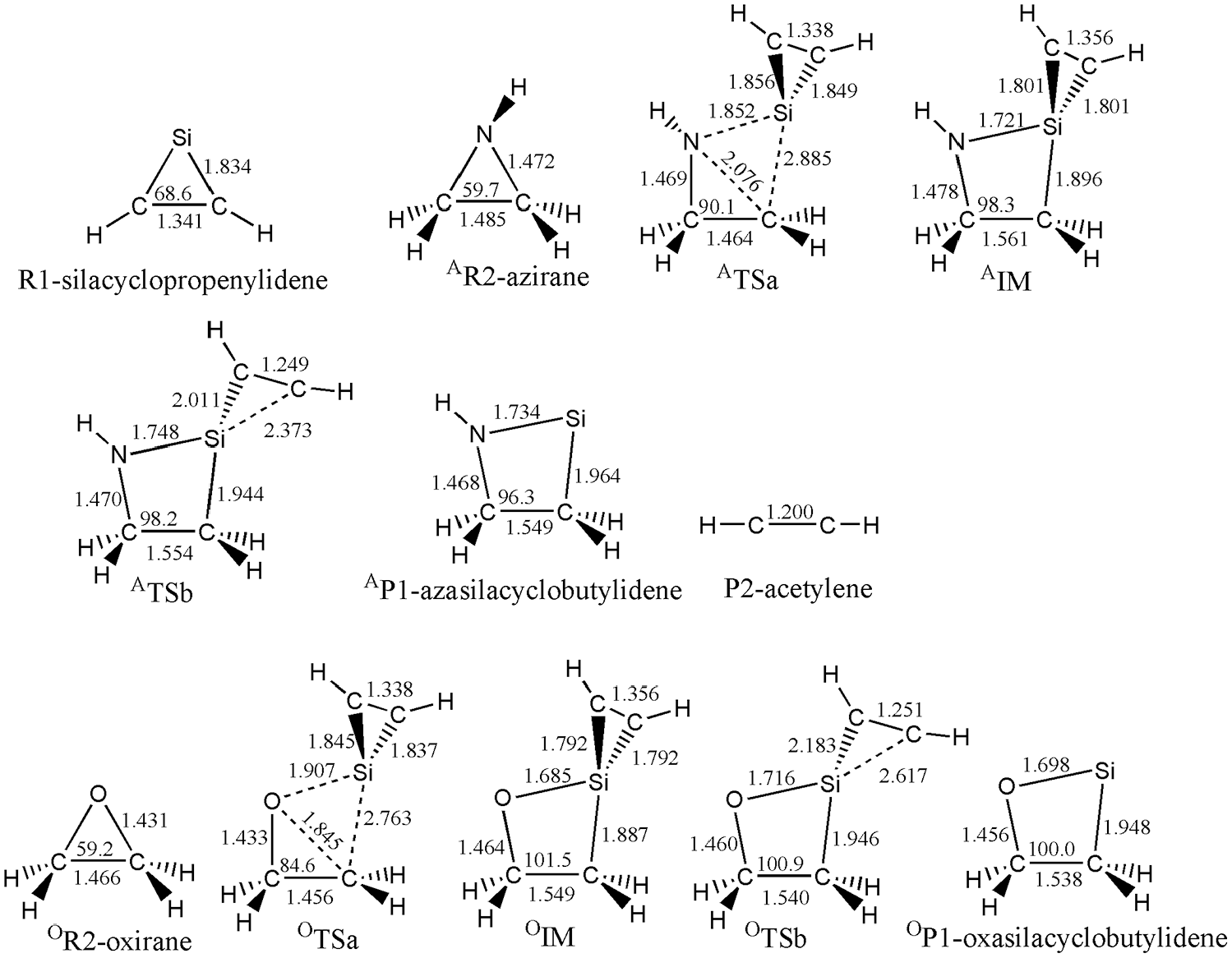
Optimized structures of the reactants (silacyclopropenylidene, and azirane or oxirane), transition states (TS), intermediates (IM), and products (azasilacyclobutylidene or oxasilacyclopropylidene, and acethylene) in the reaction at the B3LYP/6-311+G* level of theory, where the bond length and bond angle are in angstrom and degree, respectively.
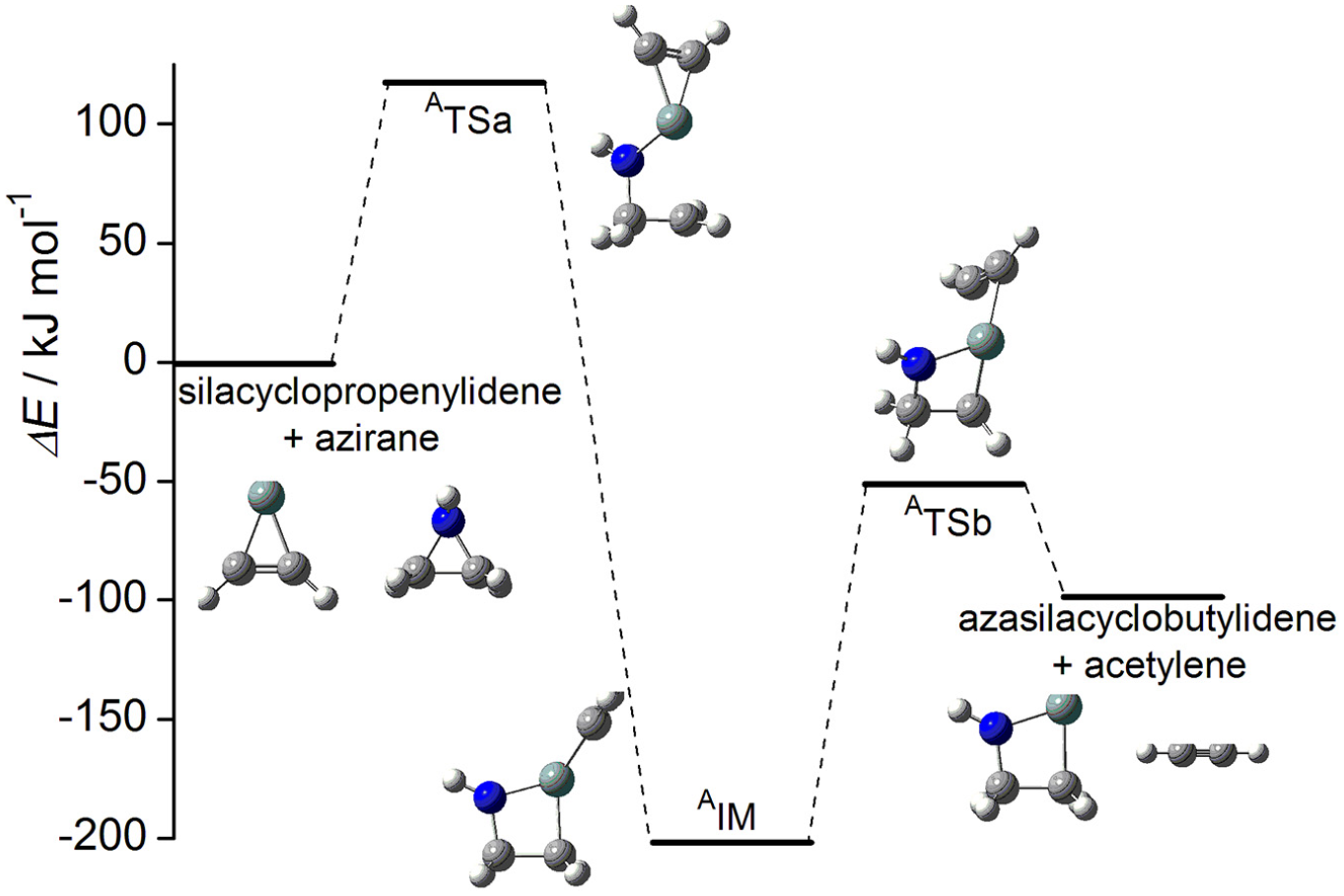
The profile for the reaction between silacyclopropenylidene and azirane at the B3LYP/6-311+G* level of theory.
Step (a): insertion process to form a Si-spiroheterocyclic intermediate IM
Along the reaction pathway of silacyclopropenylidene and azirane, the intermediate AIM is formed via an insertion process with an energy barrier of 118.1 kJ mol−1. The unique imaginary frequency calculated for the corresponding transition state, ATSa, in the step (a) is 541i cm−1 at the B3LYP/6-311+G* level of theory.
As shown in Figure 1, in ATSa, the distances of Si-N and Si-C 4 are 1.852 and 2.885 Å, respectively. Thus, two new bonds, namely Si-N and Si-C 4 , are being formed in the transition state ATSa. At the same time, the distance of N-C 4 in ATSa reached 2.076 Å, which is 0.608 Å longer than that in azirane. Based on the bond length data, the single bond N-C 4 in azirane is being ruptured in ATSa. The formation of new σ bonds of Si-N and Si-C 4 and the rupture of σ bond of N-C 4 occurred simultaneously. As shown in Figure 3, those changes of configuration and energies can be further validated by an IRC calculation on the basis of ATSa.
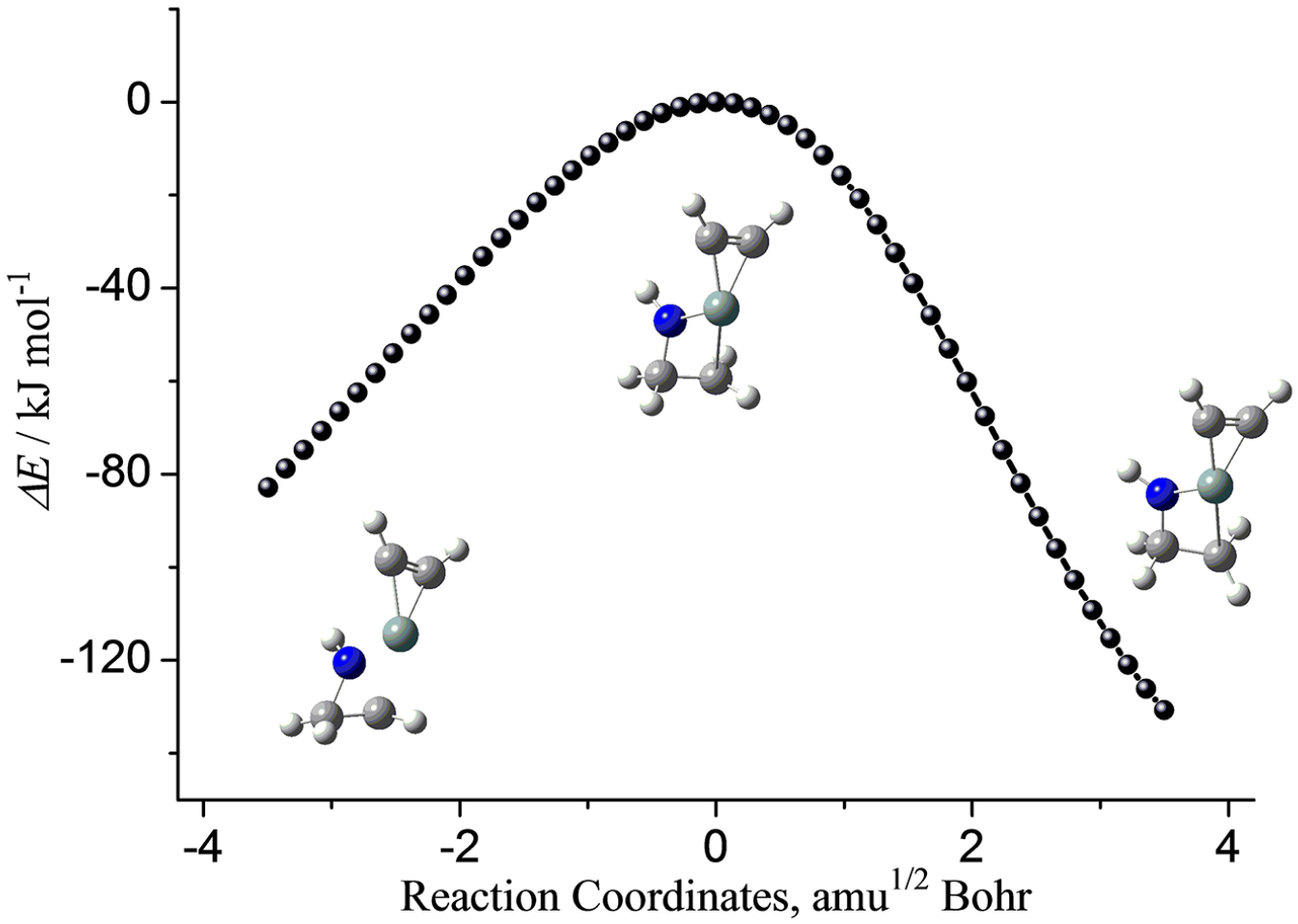
The selected configuration and energy changes along the reaction coordinate of step (a) between silacyclopropenylidene and azirane.
AIM is Si-spiroheterocyclic compound. Unlike with the singlet silylene reactant silacyclopropenylidene, all of the outer electrons of Si atom in AIM are bonded; therefore, AIM is more stable than the reactants (the energy of AIM is 199.2 kJ mol−1 lower than the reactants). However, there are two small rings in AIM. The strain in small rings is large; therefore, AIM can open its ring through rupture of the Si-C bond. As displayed in Figure 1, the intermediate is asymmetric structurally as can be reflected from the calculated C-Si bonds. As a result, the corresponding TSa is also asymmetric.
The frontier orbitals (HOMO and LUMO) of a chemical species are quite important to define their reactivity and determine the way in which the molecule interacts with other species. 36 The reaction process can be understood qualitatively from the frontier molecular orbital theory. As displayed in Figure 4, the weak strength of C-N and C-O bonds can be understood from the LUMO of azirane and oxirane, respectively. Obviously, all of them are characterized by the antibonding orbital. As for silacyclopropenylidene, the activity of Si site can be reflected from the largest contributions on the Si atom to the components of the whole HOMO. Therefore, Si atom in silacyclopropenylidene can insert into the C-N bond (in azirane) and the C-O bond (in oxirane). In addition, the LUMO of azirane is lower in energy than that of oxirane, where the orbital energies are 0.00529 and 0.01120 a.u. for the former and the latter, respectively. Therefore, the insertion process of silacyclopropenylidene with azirane is easier than that of silacyclopropenylidene with oxirane, which can be supported by the calculated barrier heights (see in Table 1).
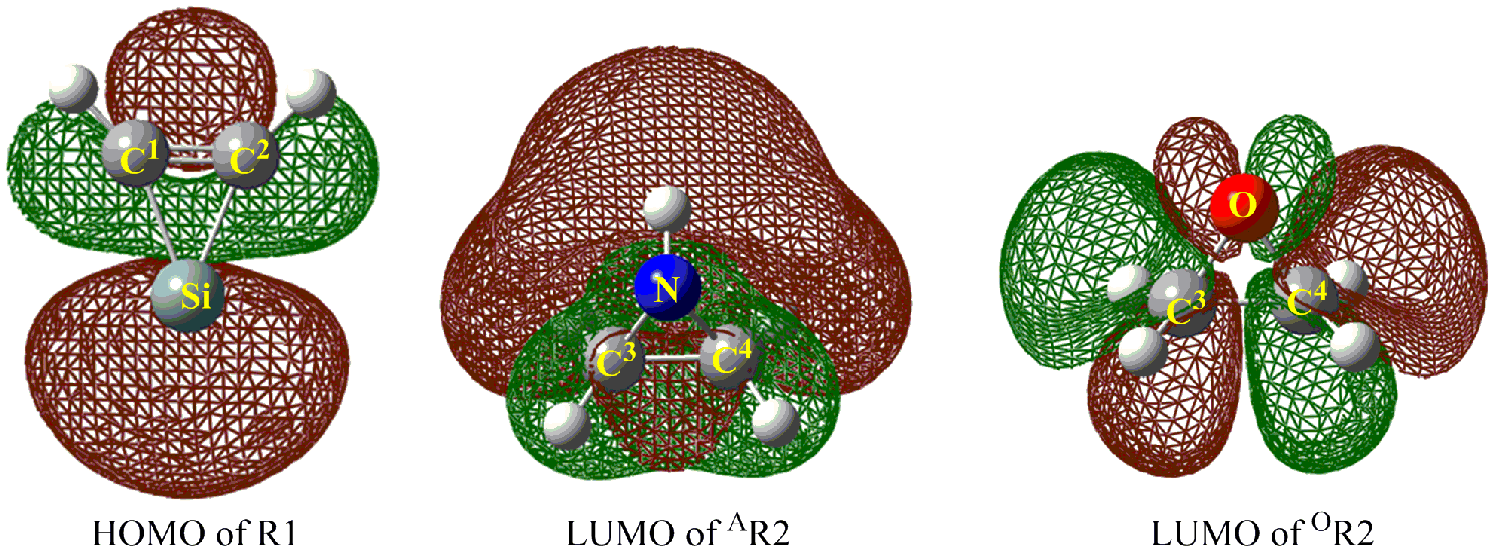
Frontier molecular orbitals of the reactants.
Because the silacyclopropenylidene uses its lone electrons to insert into C-N bond of azirane, the greater the electron density on the Si atom, the easier will be the insertion process between silacyclopropenylidene and azirane. Introduction of electron donating group in silacyclopropenylidene (c-C(H)C(Y)Si) will increase the electron density on the Si atom, which will better facilitate the insertion reaction between silacyclopropenylidene and azirane. Introduction of an electron-withdrawing group will lead to the opposite effect. As summarized in Table 1, introduction of the electron donating group, CH3, increases the electron density at Si, by which it can decrease the barrier height of step (a). On the contrary, electron-withdrawing groups, OH and OCH3, increased those barrier energies. Because the substituent group on the adjacent carbon of silicon does not much affect the electron density on the Si atom, the barrier energies changed only slightly.
The silacyclopropenylidene (c-C(H)C(H)Si) is the silicon substituted analogue of cyclopropenylidene (c-C(H)C(H)C). Silacyclopropenylidene and cyclopropenylidene have similar reactivity for this insertion process with azirane. The former’s energy barrier (118.1 kJ mol−1) is lower than the latter (210.7 kJ mol−1), 37 which demonstrated that the silacyclopropenylidene has higher reactive activity than cyclopropenylidene.
Step (b): dissociation process to form four-membered heterocyclic silylene and acetylene
As mentioned above, AIM is Si-spiroheterocyclic compound; it can open its three-membered ring (Si-C1 -C2) to form the four-membered heterocyclic silylene and acetylene through a dissociation process. Si-C1 and Si-C2 bonds in AIM will be opened to form two products via ATSb in the step (b), where the barrier is 154.3 kJ mol−1.
The unique imaginary frequency of ATSb is 379i cm−1. In ATSb, the bond lengths of Si-C1 and Si-C2 are 2.011 and 2.373 Å, which are 0.210 and 0.572 Å longer than that in the AIM, respectively, showing the bonds of Si-C1 and Si-C2 will be ruptured. Simultaneously, the bond length of C1 -C2 shortens to 1.249 Å, indicating the bond of C1 -C2 will be transferred from the double bond in AIM to triple bond in AP2. As shown in Figure 5, those changes of bond lengths and energies can be further validated by an IRC calculation on the basis of ATSb.
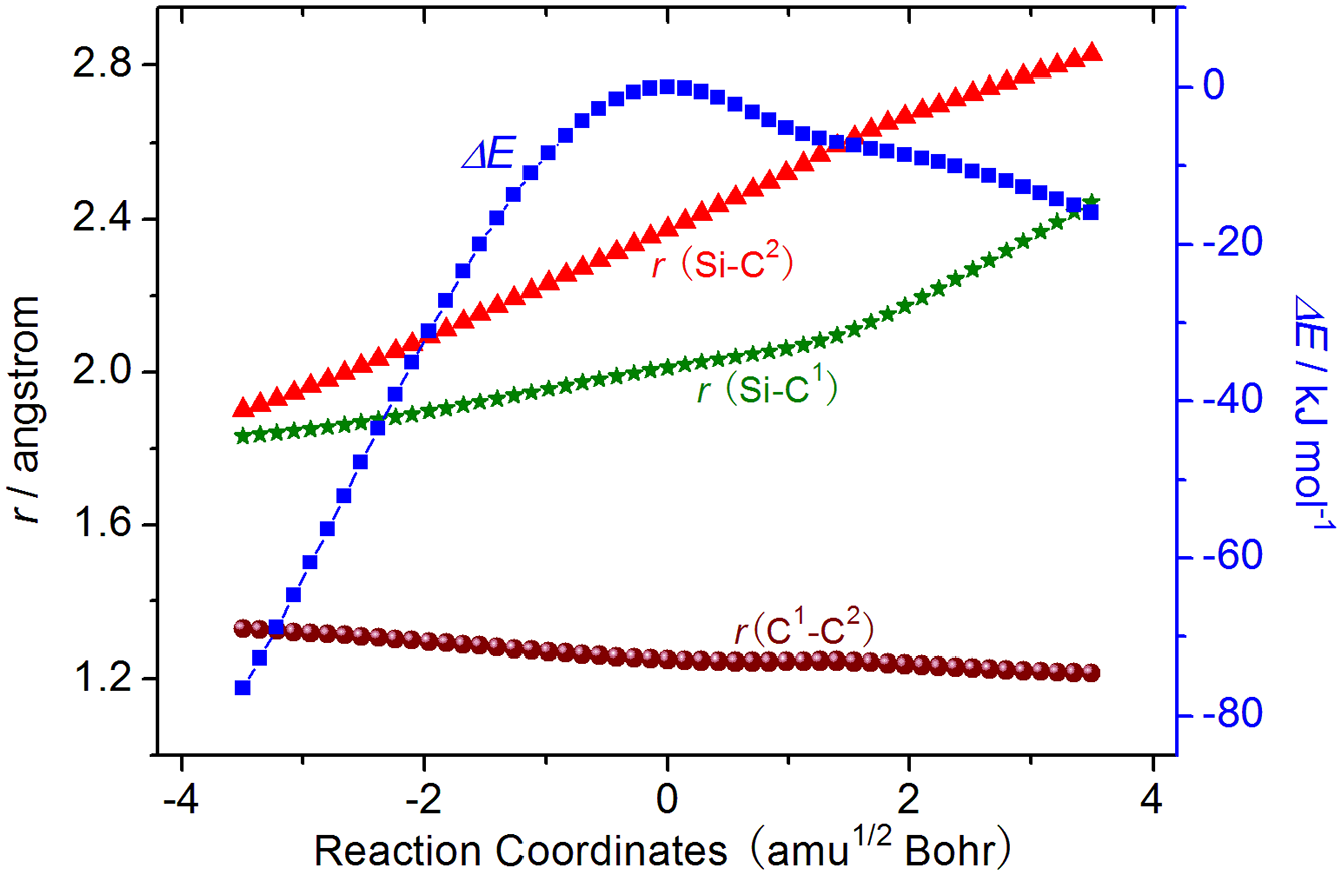
The selected bond lengths and energy changes along the reaction coordinates of step (b) between silacyclopropenylidene and azirane.
The two products of the reaction of silacyclopropenylidene and azirane are azasilacyclobutylidene and acetylene. The azasilacyclobutylidene is a four-membered heterocyclic compound. Like the reactant silacyclopropenylidene, it has the characteristics of a silylene. Azasilacyclobutylidene has not only lone pair electrons, but also has a four-membered heterocyclic ring. Therefore, azasilacyclobutylidene should be very reactive and act as a potential reaction reagent in organic chemistry. There is a similar reaction between silacyclopropenylidene and oxirane. The two products are oxasilacyclobutylidene and acetylene. Because silacyclopropenylidene might be abundant in space, the study of this reaction is useful to understand the evolution of silicon-bearing molecules in space. As well, the present study offers an alternative approach to form the active four-membered heterocyclic silylene compound.
Conclusion
In this study, the reaction mechanism between silacyclopropenylidene and three-membered heterocyclic compounds (azirane and oxirane) has been systematically investigated employing the B3LYP/6-311+G* level of theory. There are steps (a) and (b) along the reaction pathway. The Si-spiroheterocyclic intermediate was obtained through step (a). Introducing an electron donating group in the silacyclopropenylidene makes the insertion process easier, but an electron-withdrawing group produces the opposite effect. The four-membered heterocyclic silylene azasilacyclobutylidene (and oxasilacyclobutylidene) and small molecule acetylene were produced through step (b). The calculated activation energies for the step (a) (118.1 (silacyclopropenylidene and azirane) and 121.3 (silacyclopropenylidene and oxirane) kJ mol−1) are rather large; therfore, the title reactions proceed slowly in space environment. The present study is helpful to understand the reactivity of silacyclopropenylidene and the evolution of silicon-bearing molecules not only in space environment but also in laboratory environment, and to offer an alternative approach to the formation of active four-membered heterocyclic silylene compounds.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The project was funded by Project of Research Leader Studio of University Funding 20 Items in Jinan (2019GXRC058), National Natural Science Foundation of China (NSFC) (31370090), and Project of Iceland Research Center of University of Jinan (18GB04).