
Abstract
Theoretical investigations have been performed on the kinetics of bimolecular hydrogen-abstraction reactions of 1,3,5-trioxane and 1,4-dioxane cyclic ethers with OH radicals. Hydrogen abstraction from both axial and equatorial positions of 1,3,5-trioxane and 1,4-dioxane was considered. Optimization of the structures, and the calculation of energies, vibrational frequencies and moments of inertia for all the stationary points including reactants, hydrogen-bonded complexes, transition states and products were carried out using density functional theory at the M06-2X level together with the MG3S basis set. Single-point energy calculations on the optimized points were obtained at the CBS-QB3 level. The calculations show that the title reactions proceed through relatively strong hydrogen-bonded complexes due to the hydrogen bonding between the OH radicals and the oxygen atoms of the cyclic ethers. A two-transition state model (an inner tight transition state and an outer loose transition state) was employed to compute the hydrogen-abstraction rate coefficients. The rate coefficients were also computed using conventional transition state theory considering a tight transition state for the purpose of comparison. It was found that when the reactions proceed via inner transition states with relative energies higher than the reactants, the computed rate coefficients are underestimated by conventional transition state theory.
Keywords
Introduction
Ethers are an important class of compounds which are widely used as octane-enhancing additives and solvents to replace chlorocarbons and aromatic hydrocarbons. 1 1,3,5-Trioxane is consumed to produce 1 million tonnes of polyoxymethylene plastics per year. 2 The gas-phase kinetics of the reactions of cyclic ethers such as 1,3,5-trioxane (C3H6O3) and 1,4-dioxane (C4H8O2) have been studied to understand the impacts of the structures of these compounds on their atmospheric reactivity and degradation pathways.3–10 As a consequence, the kinetics of the reactions of 1,3,5-trioxane and 1,4-dioxane with OH radicals (known as atmospheric detergents) have been the subject of some experimental studies.5–10
To date, three experimental measurements of the rate coefficients for the C3H6O3 + OH reaction have been reported.5–7 Zabarnick et al. 5 have determined the rate constants of the C3H6O3 + OH reaction over the temperature range 292–597 K. By employing a laser photolysis technique for generating OH radicals and laser-induced fluorescence (LIF) for monitoring their decay under pseudo first-order conditions, they have obtained the Arrhenius equation k(T) = 1.36 × 10−11 × exp (–1.92 kJ mol−1/RT) cm3 molecule−1 s−1. A value of 6.0 × 10−12 cm3 molecule−1 s−1 at 298 K was reported by Platz et al. 6 Using a photolytic relative rate method at atmospheric pressure, Moriarty et al. 7 have obtained the Arrhenius expression k(T) = 9.6 × 10−12 × exp (–1.53 kJ mol−1/RT) cm3 molecule−1 s−1 over the temperature range 263–372 K for the reaction of C3H6O3 with OH.
The rate coefficients for the reaction of 1,4-dioxane with OH radicals have been measured by four research groups.7–10 Using the flash photolysis resonance fluorescence technique, Dagaut et al. 8 have obtained the Arrhenius expression k(T) = 8.3 × 10−12 × exp (+0.67 kJ mol−1/RT) cm3 molecule−1 s−1 for the temperature range 240–440 K. Porter et al. 9 have employed an LIF technique and obtained a value of 1.26 × 10−11 cm3 molecule−1 s−1 at 298 K. Maurer et al. 10 have performed a Fourier transform infrared kinetic and product study of the OH-radical-initiated oxidation of 1,4-dioxane in a quartz-glass photoreactor in the laboratory under different conditions and also in the outdoor EUPHORE simulation chamber in Valencia, Spain. A rate coefficient of 1.26 × 10−11 cm3 molecule−1 s−1 was determined for the reaction at 298 K under a pressure of 1000 mbar in synthetic air, which was in good agreement with previously published values. Using a photolytic relative rate method at atmospheric pressure, the Arrhenius expression k(T) = 8.3 × 10−12 × exp (+0.67 kJ mol−1/RT) cm3 molecule−1 s−1 over the temperature range 263–372 K was obtained by Moriarty et al. 7
To date, no theoretical data have been reported on the kinetics and mechanism of the hydrogen abstraction from 1,3,5-trioxane and 1,4-dioxane by hydroxyl radicals. The aim of this work was to use theoretical methods to study the kinetics of the title reactions. Here, it was attempted to compute the rate coefficients by statistical rate theories. As discussed in the next sections, the reactions proceed through a hydrogen-bonded (HB) complex following a saddle-point structure. Therefore, a two-transition state (TS) model was employed to calculate the rate coefficients.
Computational details
Electronic structure calculations
The geometries of reactants, HB complexes, TSs and products were optimized using density functional theory (DFT) at the M06-2X level, 11 together with the MG3S basis set. 12 The vibrational frequency calculations were performed using DFT at the M06-2X level together with the MG3S basis set. In order to calculate the energies more accurately, single-point energy calculations were carried out by the combination CBS-QB3 method. 13 Single-point energy calculations were also performed using the M05-2X/MG3S 14 and MPWB1K/MG3S 15 methods for comparison. M05-2X, M06-2X and MPWB1K are hybrid meta-DFT methods which were developed by Truhlar and coworkers and assessed against many reliable data sets of energies and vibrational frequencies. They were optimized to be suitable for thermochemical and kinetic calculations. All of the quantum chemistry calculations were carried out using the Gaussian 09 package. 16
Rate constant calculations
As explained in the next sections, the reactions of C3H6O3 and C4H8O2 with OH proceed through saddle-point structures before which HB complexes are formed. In each title reaction, two saddle-point structures appear. One has lower energy and the other has higher energy relative to the reactants. The negative value for the saddle-point energy indicates that, at least in the low molecular energy range, there is a second TS (critical configuration) at large separations, at which the sum of quantum states is a minimum. 17 A question arises as to which structure is the TS, the inner tight TS or the loose TS before the formation of the HB complex. A two-TS model is proposed to be a suitable approach for computing the reactive flux along the reaction path. 17 An initial long-range outer TS is considered during the formation of the HB complex. The inner TS is located around the saddle point on the potential energy surface. It is shown that at low temperatures, the outer loose TS lying at large separations beyond the HB minimum is dominant, while at high temperatures, the inner tight TS is the dominant state. Due to the negative energy of the saddle point relative to the reactants, a small negative activation energy is expected for this type of reaction.
The rate constant was computed by the standard Boltzmann average of the effective TS flux 17

where

In equation (2),
In order to compute the sum of states of the outer TS, variable reaction coordinate transition state theory (VRC-TST) was employed.18–23 In VRC-TST, which is usually applied to barrierless association processes having loose TSs, the internal degrees of freedom are subdivided into transitional modes and conserved modes. The internal modes of associating fragments whose nature changes appreciably during the association process are named as transitional modes. The remaining modes corresponding to internal vibrational modes of each fragment, whose vibrational frequencies do not change during the process, are termed conserved modes. On the basis of this subdivision of the coordinates,
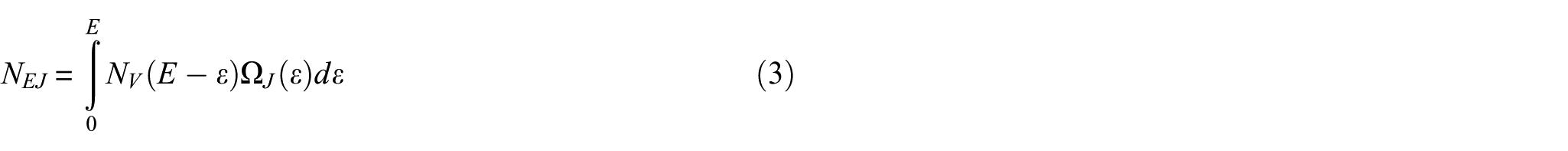
where
Results and discussion
Figures 1 and 2 show the optimized geometries of all the stationary points involved in the hydrogen-abstraction reactions of OH radicals from C3H6O3 and C4H8O2. As can be seen, OH radicals abstract the H atoms of C3H6O3 and C4H8O2 via axial and equatorial positions. The two possible pathways for hydrogen-abstraction processes were considered in the calculation of rate constants. The z-matrices for the structures of reactants, HB complexes and TSs are provided in the Supplemental material. On the basis of the energies computed at the CBS-QB3 level of theory, the title reactions can be described as follows. The reaction C3H6O3 + OH initially proceeds through a common HB complex, denoted as HB1t. The energy of the HB1t complex is 20.0 kJ mol−1 lower than that of the reactants. This complex is formed via a hydrogen bond between the hydrogen atom of the hydroxyl radical and an oxygen atom of C3H6O3. Next, hydrogen abstraction from C3H6O3 by the OH radical occurs via the TS structures TS1t and TS2t corresponding to axial and equatorial hydrogen atoms on C3H6O3. The energies of TS1t and TS2t relative to the reactants’ energy are −3.1 and 9.7 kJ mol−1, respectively. Next, TS1t and TS2t lead to two HB complexes HB2t and HB3t with relative energies of −112.0 and −91.7 kJ mol−1, respectively. In the latter complexes, the products H2O + C3H5O3 are attached to each other via hydrogen bonding interactions between an H atom of the H2O molecule and an O atom of C3H5O3. These complexes decompose to yield the corresponding products P1t and P2t, with relative energies of −74.5 and −43.7 kJ mol−1, respectively. The relative energies of the stationary points located on the potential energy surfaces of the reaction C3H6O3 + OH are illustrated in Figure 3.
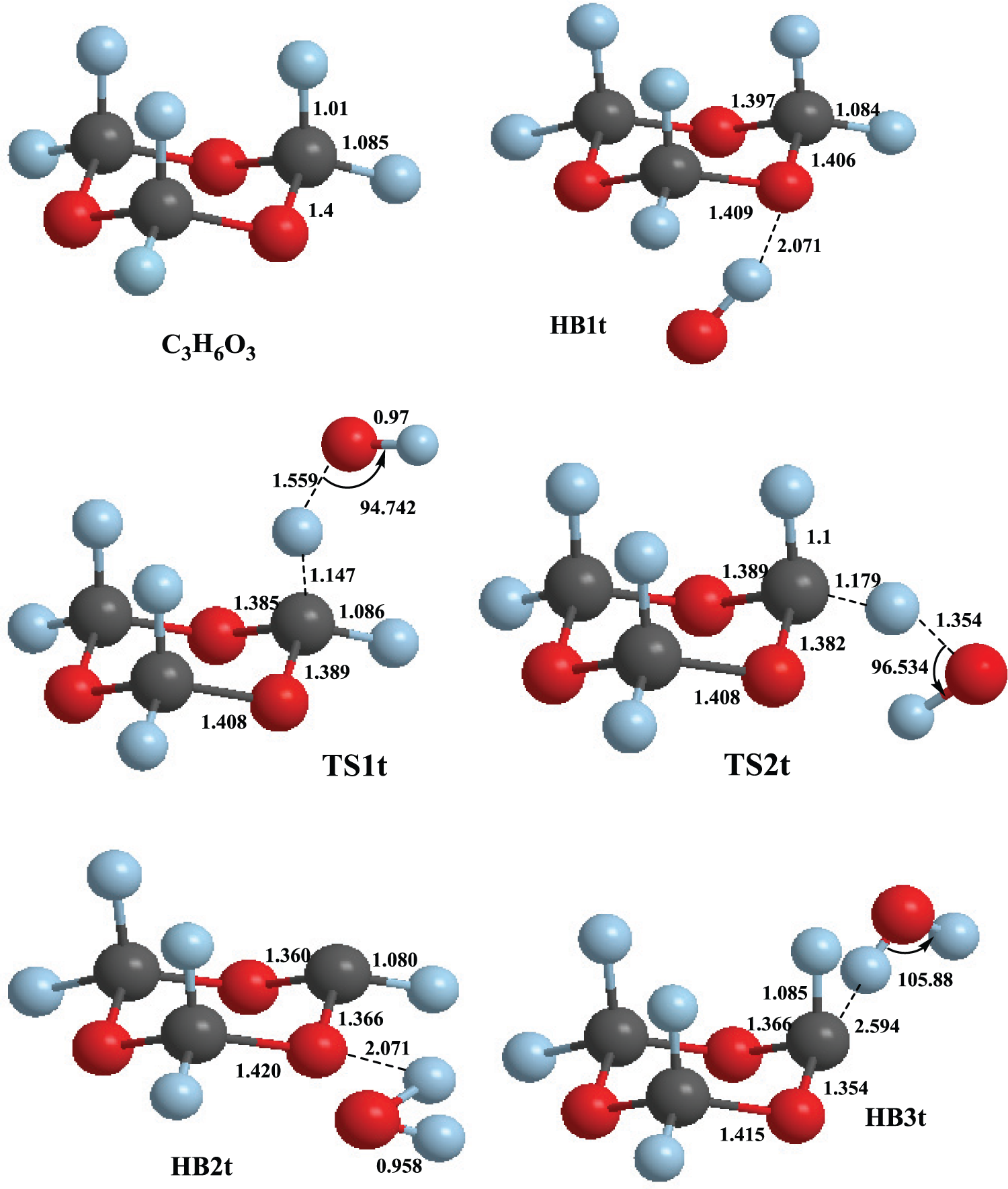
Geometries of stationary points involved in the C3H6O3 + OH reaction optimized at the M06-2X/MG3S level of theory.
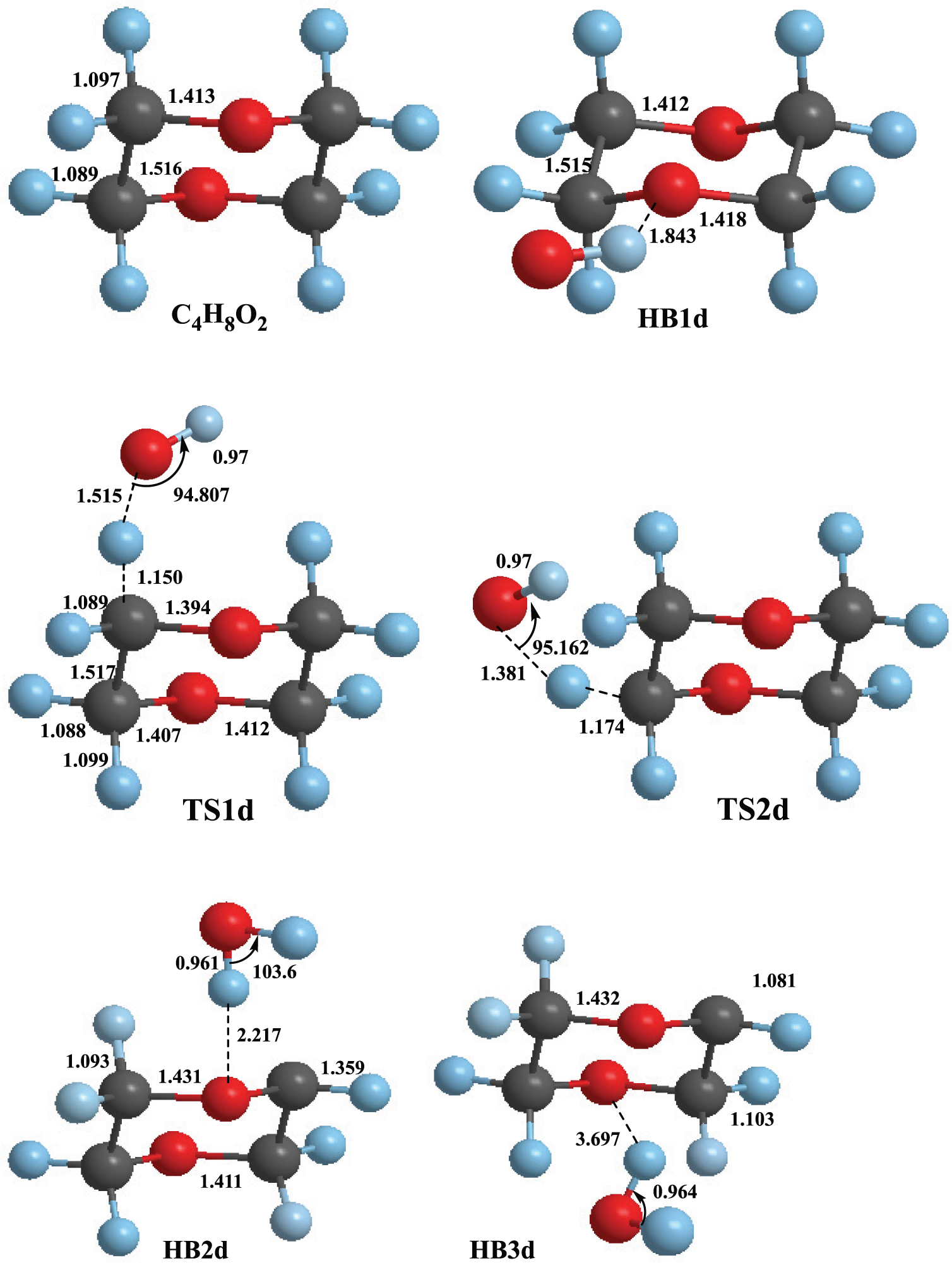
Geometries of stationary points involved in the C4H8O2 + OH reaction optimized at the M06-2X/MG3S level of theory.
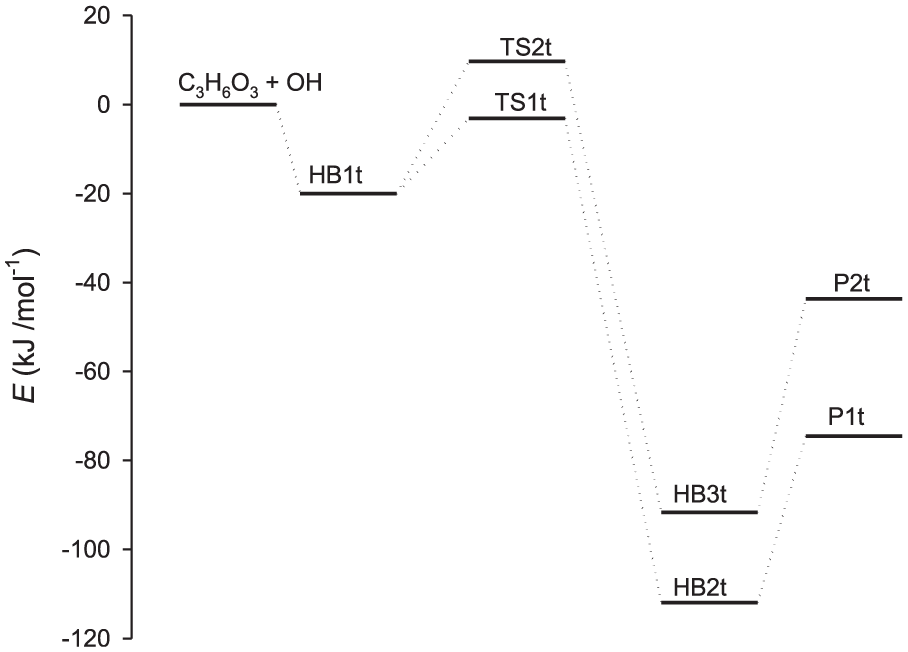
Relative energies for potential energy surfaces of the C3H6O3 + OH reaction
The reaction of an OH radical with C4H8O2 occurs with a similar mechanism. The reaction initially occurs via an HB complex, denoted as HB1d. The energy of the HB1d complex is 26.5 kJ mol−1 lower than that of the reactants. Next, hydrogen abstraction from C4H8O2 by the OH radical proceeds through the TS structures TS1d and TS2d corresponding to axial and equatorial hydrogen atoms on C4H8O2. The energies of TS1d and TS2d relative to the reactants are −1.9 and 2.4 kJ mol−1, respectively. Next, TS1d and TS2d lead to two HB complexes HB2d and HB3d with relative energies of −106.2 and −112.2 kJ mol−1, respectively. These complexes decompose to yield products P1d (C4H7O2 + H2O) with a relative energy of −89.2 kJ mol−1. The relative energies of the stationary points located on the potential energy surfaces of the reaction C4H8O2 + OH are illustrated in Figure 4.
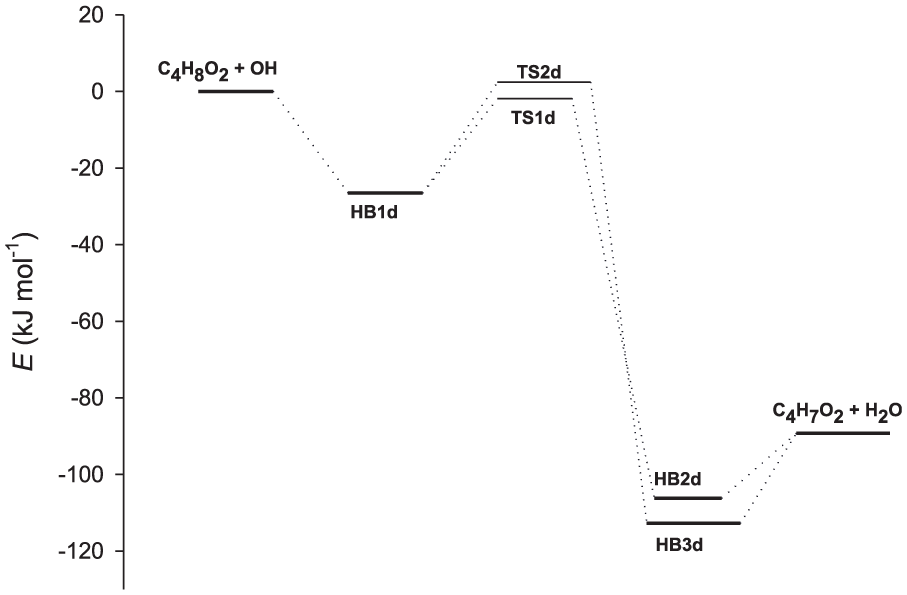
Relative energies for potential energy surfaces of the C4H8O2 + OH reaction.
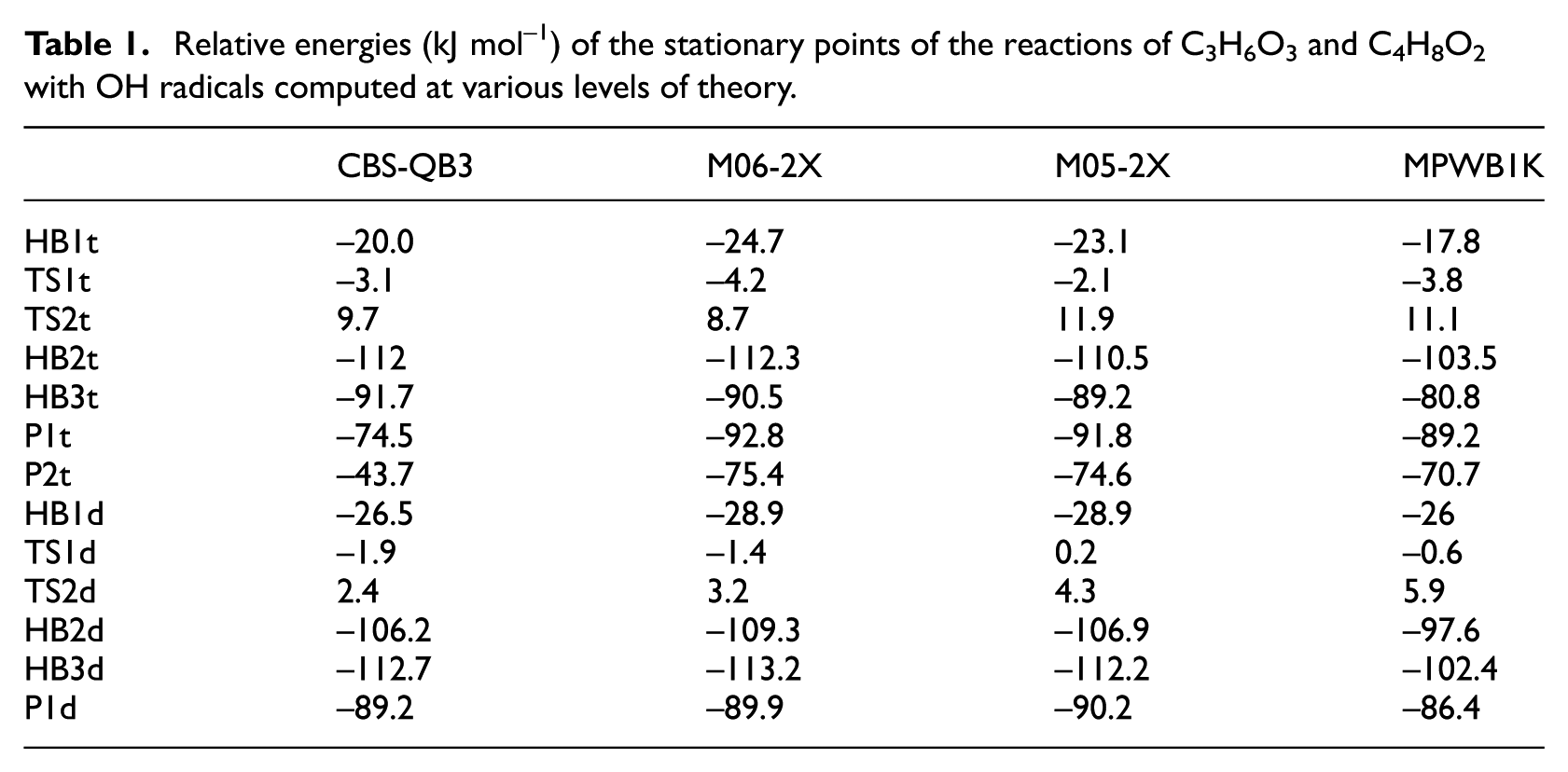
The computed energies relative to the reactants for the TSs, HB complexes, and products of the title reactions at different levels of the theory are given in Table 1. The moments of inertia and vibrational frequencies of the reactants and TSs, calculated at the M06-2X/MGS3 level of theory, are provided in Table 2S in the Supplemental material. As mentioned in the theoretical section, the outer TSs in both title reactions are considered as loose TSs, and VRC-TST is used to calculate their sums of quantum states. The sums of states for the outer TSs are computed according to equation (3). The parameters of the Varshni potential equation are obtained by fitting the Varshni equation to the computed energies at the CBS-QB3 level along the association pathway. The parameters D, re, and
Relative energies (kJ mol−1) of the stationary points of the reactions of C3H6O3 and C4H8O2 with OH radicals computed at various levels of theory.
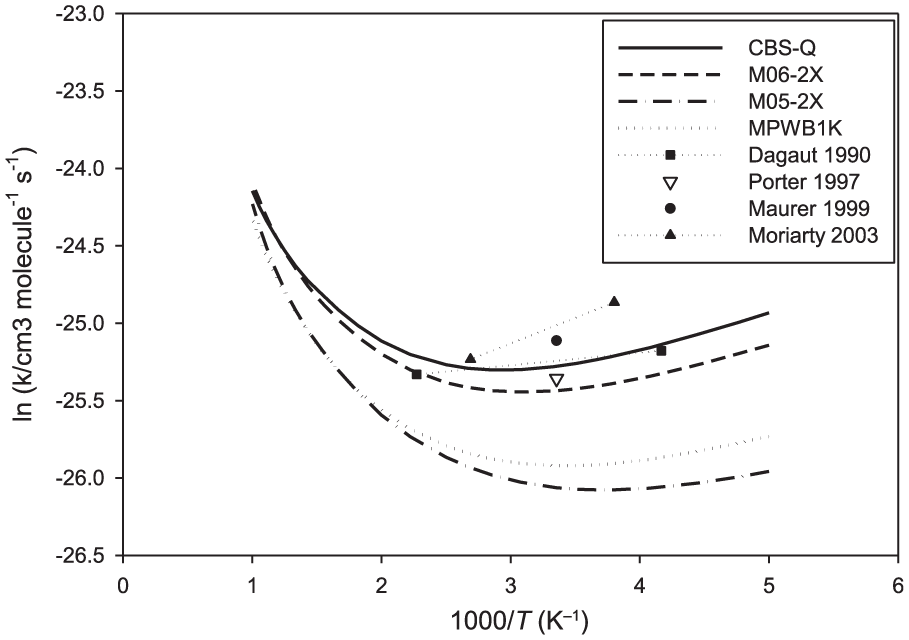
The computed rate coefficients for axial and equatorial hydrogen abstraction from 1,3,5-trioxane using CBS-QB3 energies are depicted in Figure 5. The computed rate coefficients using conventional transition state theory (CTST) are also given for the purpose of comparison (red lines). As expected, the reaction mainly proceeds via the abstraction of the axial hydrogen of C3H6O3 (via TS1t) due to the lower barrier height of this reaction channel. It is seen that the computed rate constants for the equatorial hydrogen abstraction are underestimated by CTST. The rate constants computed by CTST for the axial hydrogen abstraction are nearly the same as those computed by the two TS model. The overall rate constants for the C3H6O3 + OH reaction, computed over the temperature range 200–1000 K, are illustrated in Figure 6. The experimental values reported in the literature are also given for comparison. On the basis of the CBS-QB3 energies, the computed overall rate coefficients are between 4.6 × 10−12 and 1.3 × 10−11 cm3 molecule−1 s−1, showing a small temperature dependence. The present calculations predict a slight negative activation energy (–2.1 kJ mol−1) over the temperature range 200–300 K. The rate coefficients are predicted to be nearly constant over the temperature range 300–500 K (about 4.6 × 10−12 cm3 molecule−1 s−1), and the reactions have a positive activation energy over the temperature range 500–1000 K (6.8 kJ mol−1). From the numerical standpoint, the experimental values reported in the literature are in good agreement with the present theoretical rate coefficients using different quantum chemical methods. The computed rate coefficients using CBS-QB3 energies are in better agreement with the most recent studies by Platz et al. 6 and Moriarty et al. 7 The computed rate coefficients using M06-2X energies are close to the values reported by Zabarnick et al. 5 The numerical values for the rate coefficients of the C3H6O3 + OH reaction, for some selected temperatures, are provided in Table 2.
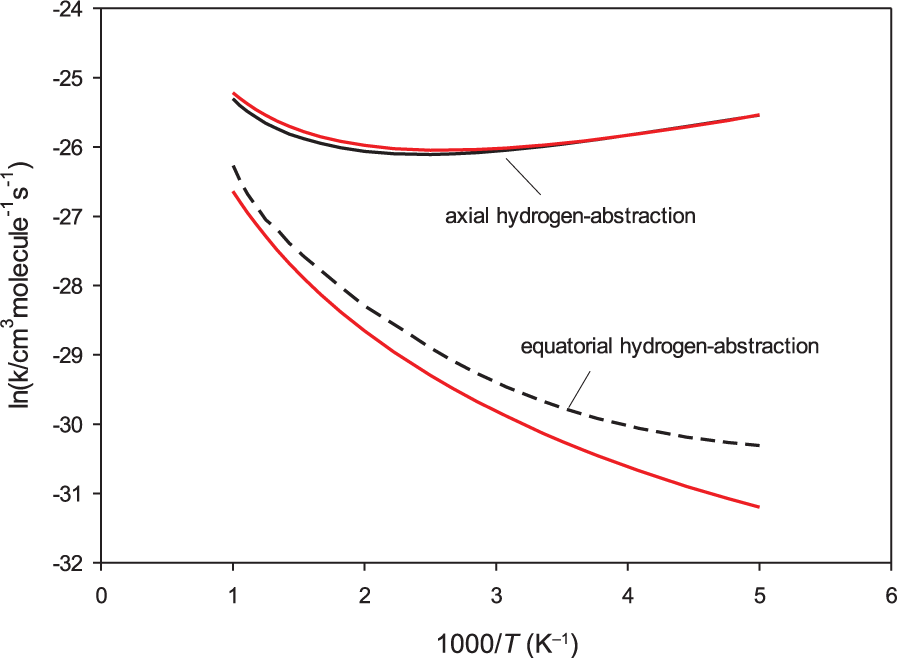
Computed rate coefficients for the hydrogen-abstraction reactions from axial and equatorial positions of 1,3,5-trioxane by OH radicals. Red lines are the computed values according to CTST.
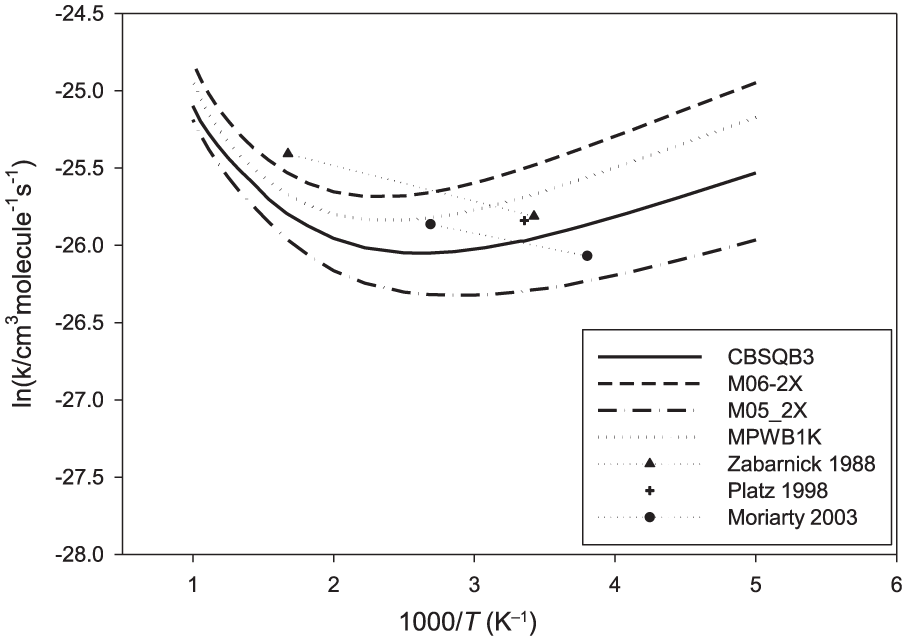
Thermal overall rate coefficients for the C3H6O3 + OH reaction computed at temperatures in the range 200–1500 K.
Computed rate coefficients (cm3 molecule−1 s−1) at some selected temperatures for the reactions C3H6O3 + OH and C4H8O2 + OH.
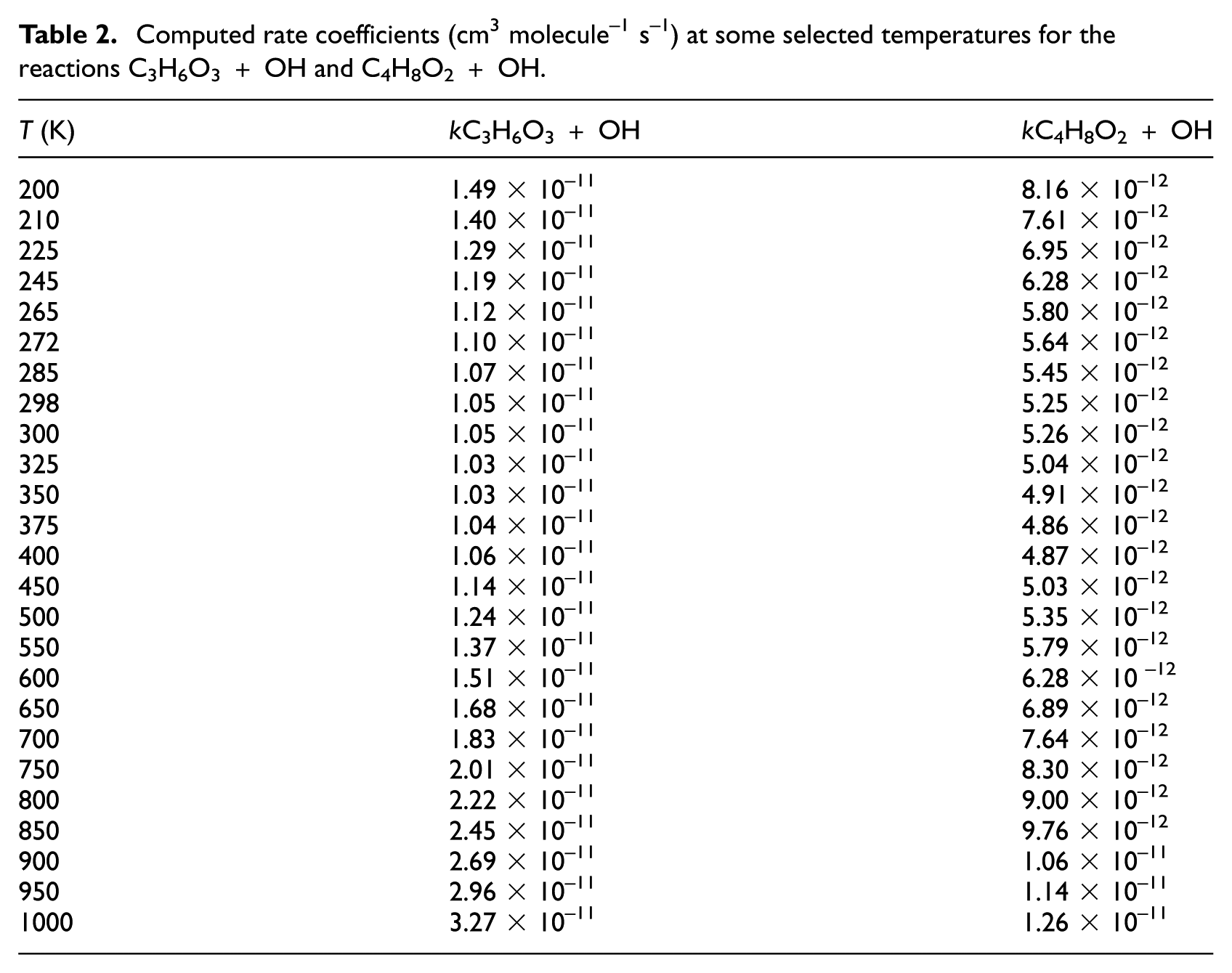
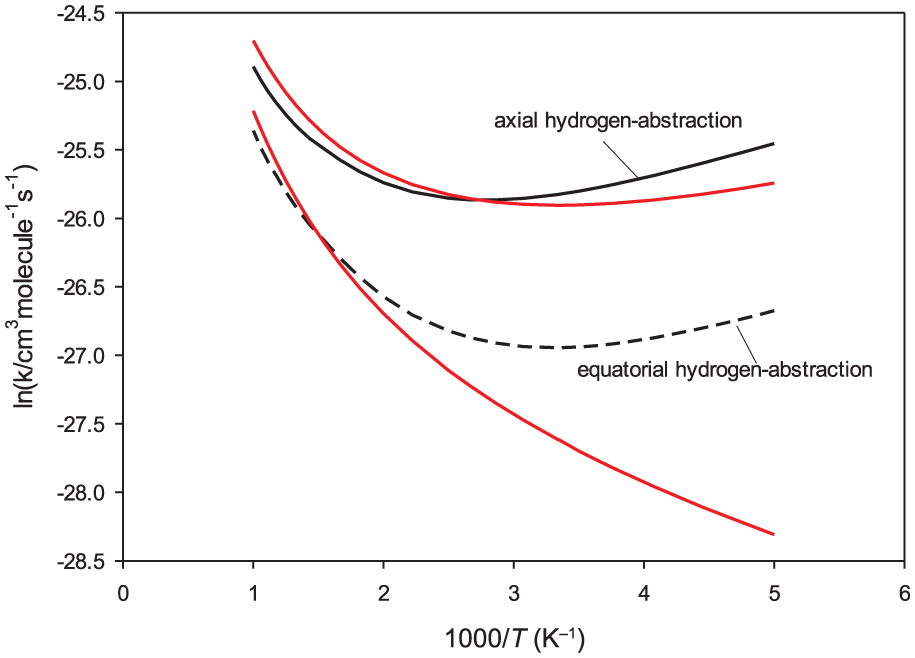
The computed rate coefficients for axial and equatorial hydrogen abstraction from 1,4-dioxane using CBS-QB3 energies are depicted in Figure 7. The rate coefficients computed using CTST are also given for comparison (red lines). As for the C3H6O3 + OH reaction, this mostly proceeds via the abstraction of the axial hydrogen of C4H8O2 (via TS1d) because the barrier height of this reaction channel is slightly lower than that of the reaction channel proceeding through TS2d. It is seen that the computed rate constants for both hydrogen-abstraction pathways are underestimated by CTST at lower temperatures. The overall rate constants for the C4H8O2 + OH reaction, computed over the temperature range 200–1000 K, are illustrated in Figure 8. The experimental values reported in the literature are also given for comparison. Essentially, similar results as for the C3H6O3 + OH reaction are obtained. On the basis of CBS-QB3 energies, the computed overall rate coefficients are between 1.0 × 10−11 and 3.4 × 10−11 cm3 molecule−1 s−1, showing a small temperature dependence. The present calculations predict a slight negative activation energy (–1.7 kJ mol−1) over the temperature range 200–300 K. The rate coefficients are predicted to be nearly constant over the temperature range 300–450 K (about 1.0 × 10−11 cm3 molecule−1 s−1), and the reactions have a positive activation energy over the temperature range 450–1000 K (7.2 kJ mol−1). The numerical values of the computed rate constants employing different quantum chemical methods show that they are in good agreement with experimental values reported in the literature. However, the computed rate coefficients using CBS-QB3 energies have the best accordance with experimental data.7–10 The numerical values for the rate coefficients of the C4H8O2 + OH reaction, for some selected temperatures, are provided in Table 2.
Computed rate coefficients for the hydrogen-abstraction reactions from axial and equatorial positions of 1,4-dioxane by OH radicals. Red lines are the computed values according to CTST.
Thermal overall rate coefficients for the C4H8O2 + OH reaction computed at temperatures in the range 200–1500 K.
The results obtained in this study are similar to those obtained for the reaction C2 H4 + OH. 17 The present calculations reveal how the structures of the cyclic ethers could affect their reactivity. Here, two important points in the computed results should be considered. First, there are two positions of cyclic ethers for hydrogen abstraction, that is, axial and equatorial, which should be distinguished. The present calculations reveal that the axial hydrogen atoms are more easily abstracted by OH radicals. A two-TS model for computing the rate coefficients of these reactions leads to results in better agreement with experimental data, especially at low temperatures. A simple CTST model, considering only the inner tight TS, usually underestimates the rate coefficients.
The tropospheric lifetimes of C3H6O3 and C4H8O2 can be estimated by comparison with the accepted value of the lifetime of methyl chloroform according to the following relationship

where τi and τCH3CCl3 are the tropospheric lifetimes of the cyclic ether and CH3CCl3, respectively, and ki and kCH3CCl3 are the rate coefficients for the reactions of the cyclic ether and CH3CCl3 with OH radicals at 272 K, respectively. The accepted values for kCH3CCl3 at 272 K and τCH3CCl3 in the literature are 6.0 × 10−15 cm3 molecule−1 s−1 and 5.99 years, respectively.24,25 The atmospheric lifetimes for C3H6O3 and C4H8O2 are estimated to be 29 and 56 h, respectively.
Conclusion
Here, the rate coefficients for the hydrogen-abstraction reactions of 1,3,5-trioxane and 1,4-dioxane by OH radicals have been computed by statistical rate theories. The DFT method M06-2X/MG3S was employed to optimize the structures of the stationary points of the reactions. In order to have better estimations of energies, single-point energy calculations were also performed at the CBS-QB3 level of theory. It was found that both title reactions proceed through initial relatively strong HB complexes. A two-TS model was employed to compute the rate coefficients over the temperature range 200–1000 K. The computed rate constants were in good agreement with available experimental data. It was concluded from the computed results that a long-range loose outer TS is the bottleneck of the reaction at low temperatures, while the inner tight TS is the bottleneck at high temperatures. On the basis of the present calculations, the atmospheric lifetimes of C3H6O3 and C4H8O2 were predicted to be 29 and 56 h, respectively.
Supplemental Material
PRK1800738v2_ESI – Supplemental material for Theoretical studies on the kinetics of the hydrogen-abstraction reactions from 1,3,5-trioxane and 1,4-dioxane by OH radicals
Supplemental material, PRK1800738v2_ESI for Theoretical studies on the kinetics of the hydrogen-abstraction reactions from 1,3,5-trioxane and 1,4-dioxane by OH radicals by Vahid Saheb and Aidin Bahadori in Progress in Reaction Kinetics and Mechanism
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.