
Abstract
In this article, hybrid functional B3LYP method is used to construct the reactant structure of energetic components in propellant at the bhandhlyp/6-31g(d) level, and to calculate the closed-shell layer of the system. At the bhandhlyp/6-31g(d) level, the energy difference (activation energy) between the transition state and the reactant was calculated and the reaction mechanism between energetic components was analyzed. It is found that the O30 atom of RDX first breaks off from the nitro group and is easier to break away from RDX and interact with the vertex atom Al1 of the Al13 cluster. With the further separation of O30, it also acts with Al11 until it completely breaks away from N26 atom. The activation energy of this reaction is 56.448 × 103 J mol−1. The oxygen dioxide atom in ammonium perchlorate is more likely to interact with the Al11 atom of the Al13 cluster. With the reaction proceeding, the O22 atom will not completely separate from the Cl19 atom. The activation energy of the reaction is 27.830 × 103 J mol−1.
Keywords
Introduction
Study on thermal decomposition reaction mechanism of composite solid propellant is the basis of safety performance, compatibility, and life prediction evaluation in production, transportation, and storage. 1 At present, hydroxyl-terminated polybutadiene (HTPB) composite solid propellant is widely used in many kinds of ammunition engines. Therefore, it is of great significance to study the mechanism of thermal decomposition.
Quantum chemistry has been widely used in the study of thermal reaction mechanism. Liu et al. 2 used quantum chemistry method to optimize and calculate the stable aggregate configuration of isopropyl nitrate. The static properties of isopropyl nitrate were analyzed based on this configuration. Wang and You. 3 measured the thermal decomposition process of pazufloxacin mesilate (PZFX) in nitrogen and air by TG and DSC. The bond order of PZFX molecule was calculated by quantum chemical GAMESS software, and the thermal decomposition mechanism of PZFX was deduced. In this article, a suitable density functional theory (DFT) calculation method is selected by means of quantum chemistry. According to the size of the propellant molecular system, the equilibrium geometrical structure of Al, RDX, and ammonium perchlorate (AP) molecules in the propellant was optimized, and the thermal decomposition reaction mechanism was constructed, and the activation energy between energetic components was calculated.
Thermal decomposition test
Sample
The main components of HTPB/RDX/Al/AP four-component propellant are HTPB, aluminum (Al), RDX, and AP. Other components include curing agent isophorone diisocyanate (IPDI) and plasticizer dioctyl sebacate (DOS).
Instruments and test conditions
Test instruments: Pyris-1 Thermogravimetric Analyser, PE Company, USA. Test conditions: high-purity nitrogen (99.999%), pressure of 0.3 MPa, gas flow rate of 20 mL min−1, common aluminum pool curling; heating rate of 2.5°C min−1, 5°C min−1, 10°C min−1, and 15°C min−1, respectively, from 100°C to 450°C.
Results and analysis
It can be seen from Figures 1 and 2 that the thermal decomposition weight loss of propellant can be divided into three stages. The weight loss is about 20% of the mass in the range of 150°C–220°C. The reaction rate of thermal decomposition is the fastest and the duration of thermal decomposition is shorter. The weight loss of the propellant is about 50% of its mass in the range of 220°C–375°C, during which the propellant continues to decompose at a relatively uniform rate. The weight loss is about 20% in the range of 375°C–515°C. Thermal decomposition ends after this stage. The final total weight loss is about 90% of the mass, leaving about 10% of the residue. Combined the results of DSC, the first stage is mainly the thermal decomposition weight loss of RDX. At the same time, Part of Al will react with RDX by redox reaction. RDX and AP, as strong oxidants, will react with HTPB rubber by post-curing and strong oxidation, and the main products are carbon-containing gas-phase oxides.4,5 The second stage is the thermal decomposition of AP. The third stage is high temperature decomposition of AP after transformation of crystal form 6 and thermal decomposition of HTPB rubber. However, the first stage of thermal decomposition of propellant is about 30°C ahead of RDX, and the second stage of thermal decomposition is about 35°C ahead of AP. The thermal decomposition reaction of HTPB rubber cured by isocyanate crosslinking with RDX or AP does not advance the thermal decomposition temperature of RDX or AP. 7
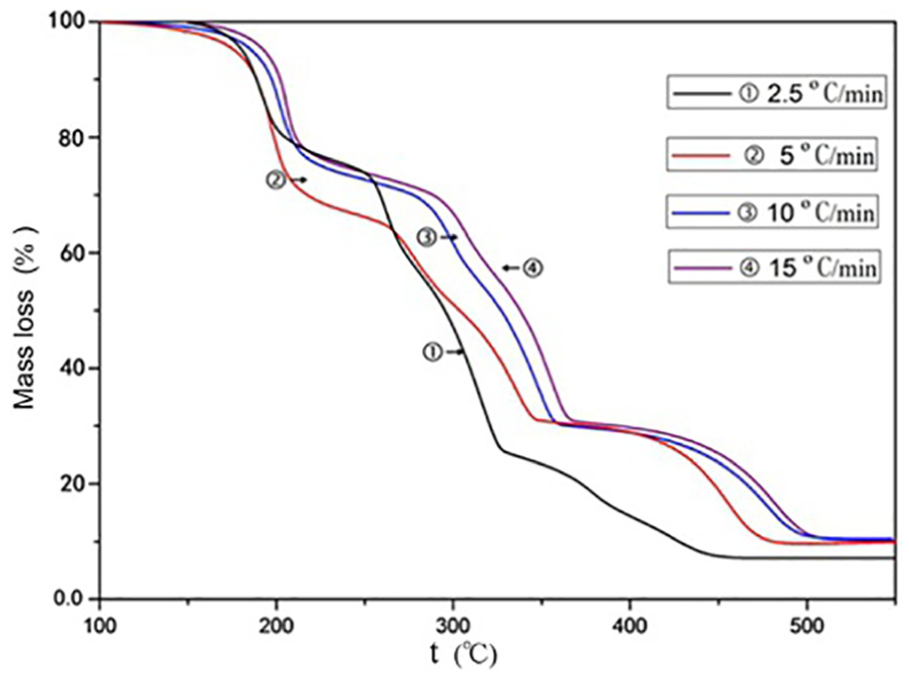
TG diagram of propellant.
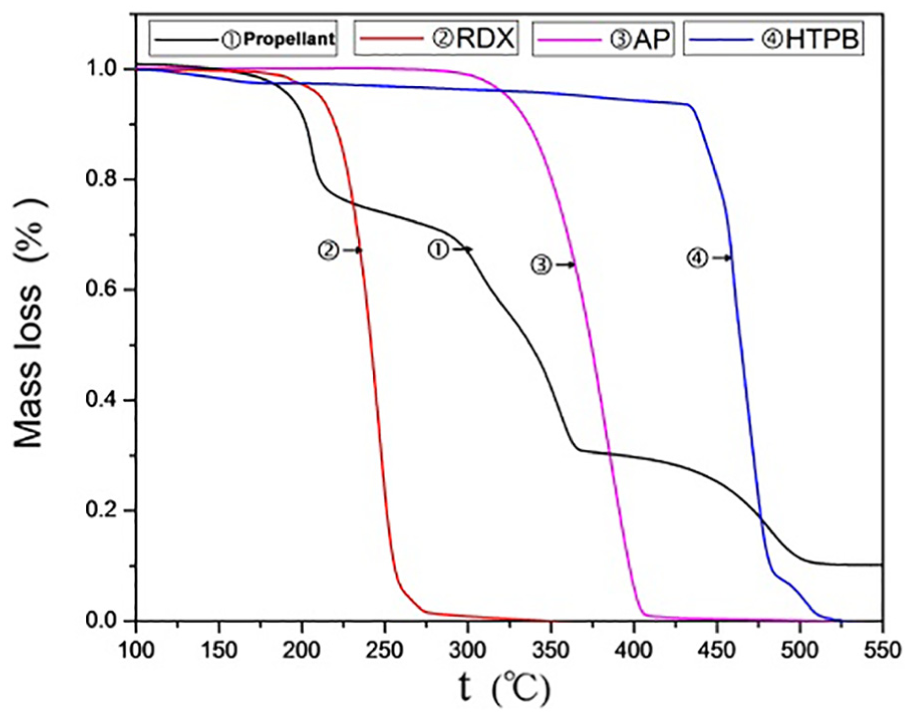
Propellant and its composition TG curve.
Quantum chemical computation
It can be seen from the experiments that there are reactions among energetic components of propellant, which lead to the earlier decomposition weight loss of components in each stage of propellant decomposition. Quantum chemistry method is used to further study the mechanism of the reactions among components and calculate the activation energy of the reactions.
All geometric structures and energy calculations are performed by Guassian 09 software. Using the hybrid functional B3LYP method in DFT, which combines Becke’s three-parameter hybrid functional with LYP correlation function, the structure of energetic component reactants in propellants is constructed at the level of bhandhlyp/6-31g(d), and the geometrical structure of reactants, products, and transition states is optimized and the vibration frequency is calculated. IRC is used to connect reactants, transition state, and products to confirm whether the transition state is correct. The closed-shell calculation is carried out for the system, and the energy difference between the transition state and reactants (i.e. activation energy) is calculated at the bhandhlyp/6-31g(d) level.
Reactant structure
The energetic components of propellant are RDX, AP, and Al. When RDX and AP are oxidants and Al is reductant, the reactions between energetic components of propellant are mainly RDX and Al, and the reactions between AP and Al. Because aluminum is a metal element, the aluminum atom has 13 extra-nuclear and the number of outermost electrons is 3. In order to fully reflect the structure and characteristics of aluminum and take account of the calculation time, this article intends to take the Al13 cluster (represented by Al below) as the main object of study. The crystal form of RDX used in this propellant is α type. A molecular model of alpha-type RDX was established. Due to the existence of hydrogen bonds (N-H···O) in NH4ClO4 crystals, this weak electrostatic interaction has an important influence on the structure and properties of NH4ClO4. 8 The calculation results will be affected. Therefore, the model of AP ionic crystal is established. The geometric configurations of the stationary points in the reactants were optimized at the bhandhlyp/6-31g(d) level. The optimum geometrical structure of each stationary point in the three reactants is shown in Figure 3. The corresponding structure information including key length and bond angle is obtained as shown in Table 1.
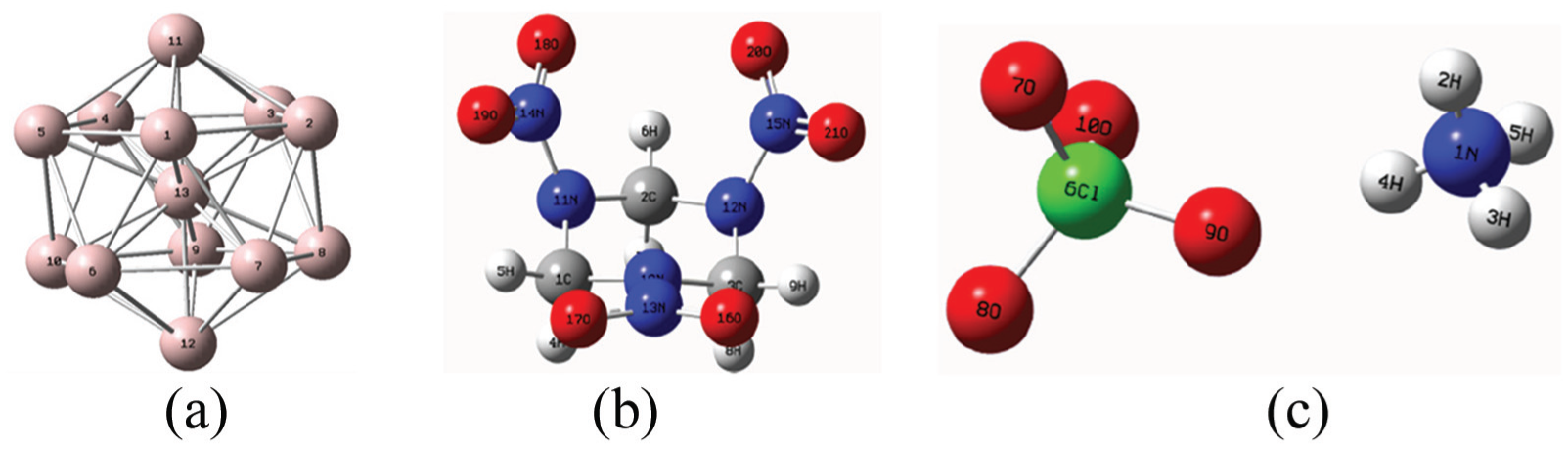
Structural diagram of three substances: (a) Al13, (b) RDX, and (c) AP.
Geometric parameters of all stationary points.
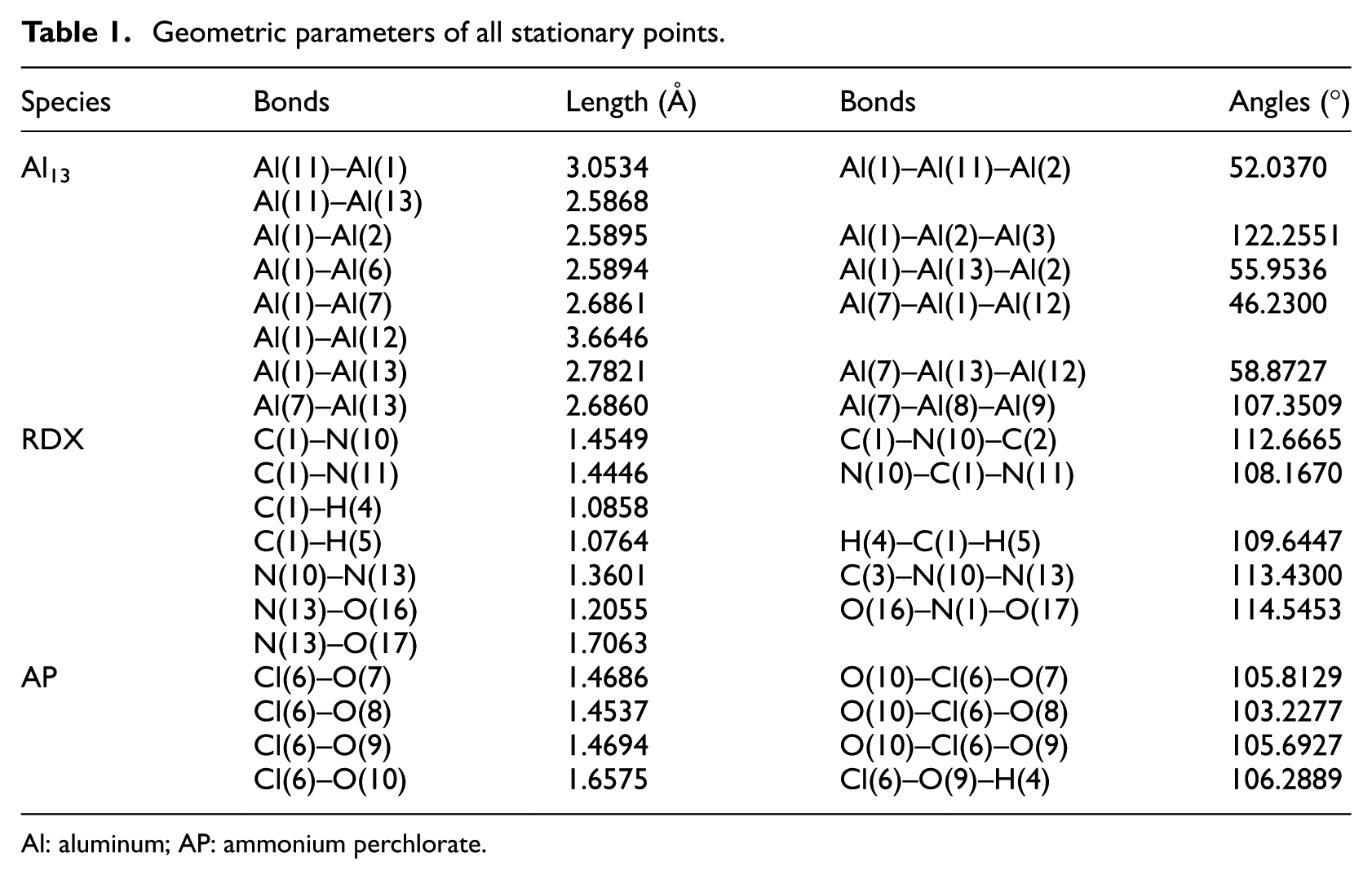
Al: aluminum; AP: ammonium perchlorate.
The structure of Al13 cluster is a regular triangular icosahedron with high Ih symmetry. Its ground state structure is mainly located near the ground state of potential energy surface and has high dynamic stability. It is easy to form a more stable structure of Al13− with good catalytic activity. 9
Establishment of reaction model
If RDX reacts with Al or AP reacts with Al, it must first react with the outermost atom of Al13 cluster, and then it is possible to react with the inner Al atom as the reaction proceeds. By analyzing the structure of Al13, we can see that there are only two kinds of atoms on the surface of Al13 clusters, which can be represented by Al1 and Al11, respectively. Because the crystal form of the RDX is α type, it can be divided into two types according to the position angle of the nitro group relative to the ring. The dihedral angle between the nitro group formed by O(29)–N(26)–O(30) and the parent ring is 157.0047°, and the dihedral angle between the nitro group formed by O(31)–N(27)–O(32) and the parent ring is −142.0121°. Therefore, four kinds of Al + RDX structures are constructed according to the structural characteristics of RDX and Al13 clusters. As shown in Figure 4,
The nitro group composed of O(29)–N(26)–O(30) reacts with Al1 to form a bond.
The nitro group consisting of O(29)–N(26)–O(30) reacts with Al11 to form a bond.
The nitro group consisting of O(31)–N(27)–O(32) reacts with Al1 to form a bond.
The nitro group consisting of O(31)–N(27)–O(32) reacts with Al11 to form a bond.
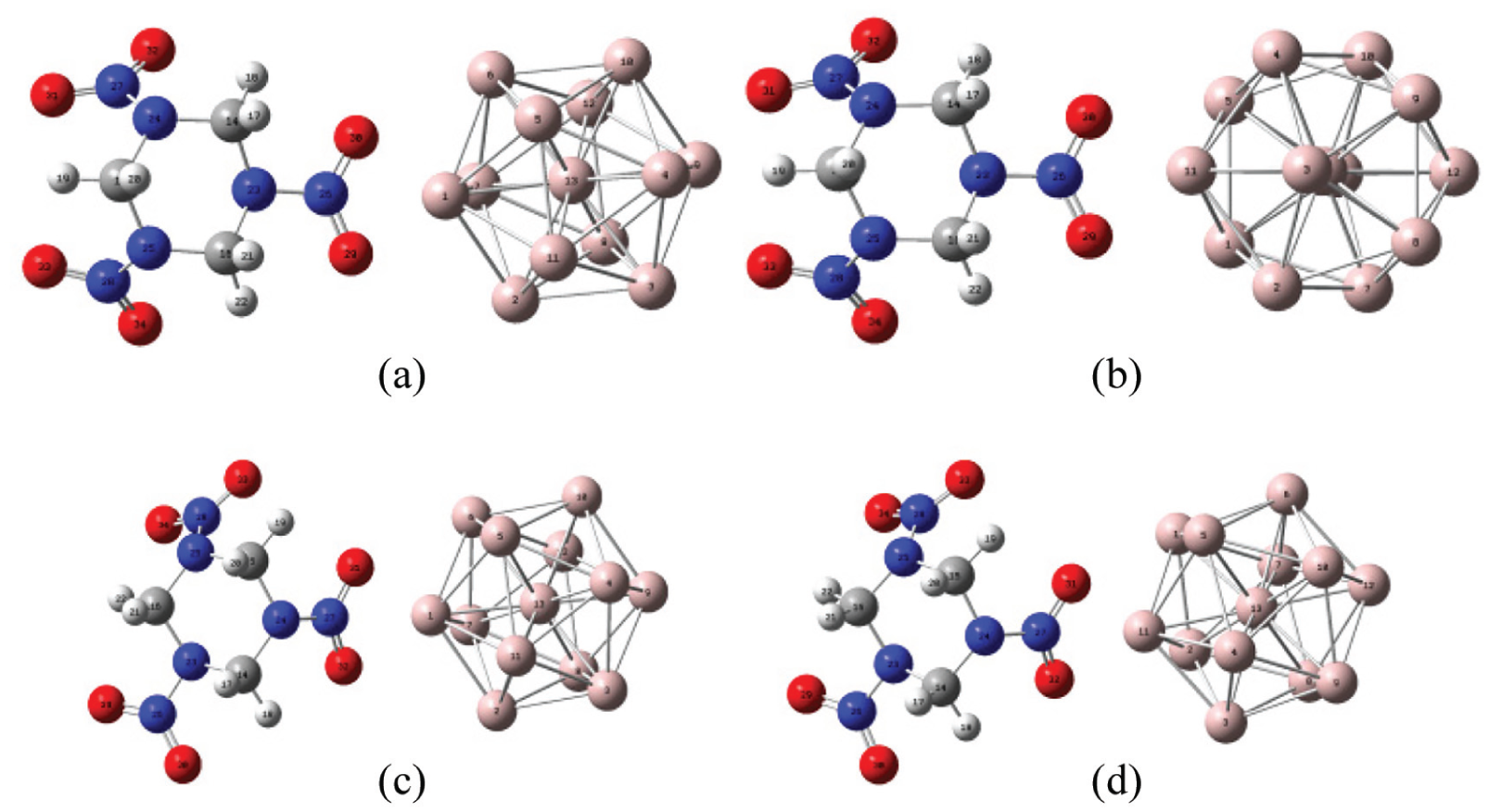
Four Al + RDX structures: (a) Structure I, (b) Structure II, (c) Structure III, and (d) Structure IV.
If AP reacts with Al, to differing positions of the NH4 counterion relative to the interacting O–Al axis, the electrostatic interaction between the NH4 counterion and oxygen atoms are also different. Eight Al + AP structures are constructed. The O atoms on HClO4− react with Al1 and Al11 to form bonds, respectively, as shown in Figure 5:
Cl(19)–O(20) reacts with Al1 or Al11 to form bonds.
Cl(19)–O(21) reacts with Al1 or Al11 to form bonds.
Cl(19)–O(22) reacts with Al1 or Al11 to form bonds.
Cl(19)–O(23) reacts with Al1 or Al11 to form bonds.
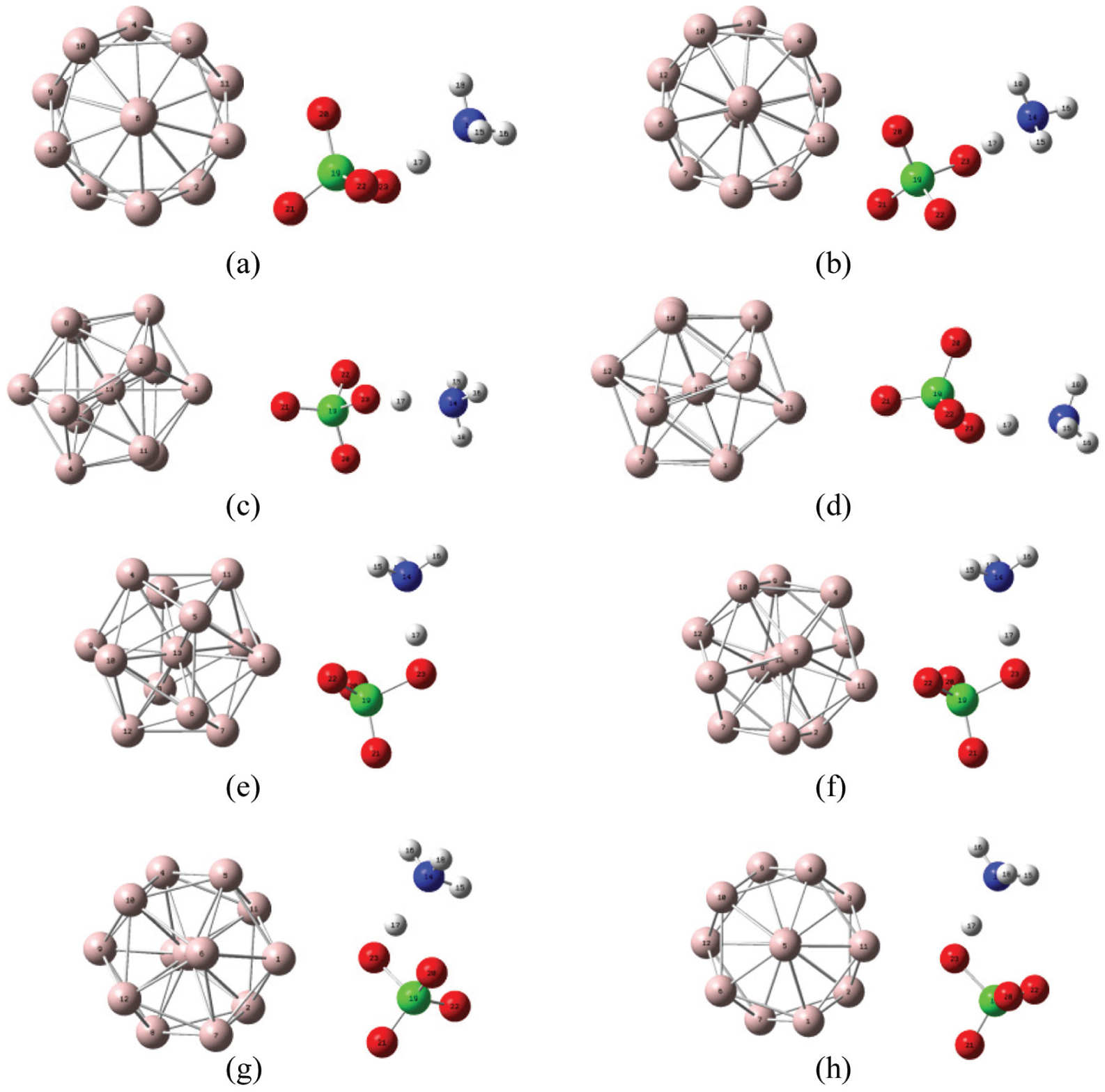
Eight Al + AP structural diagrams: (a) Structure I, (b) Structure II, (c) Structure III, (d) Structure IV, (e) Structure V, (f) Structure VI, (g) Structure VII, and (h) Structure VIII.
Reaction mechanism of RDX and Al
According to the above four reaction paths, the transition states of each path are determined, and the geometrical structures of reactants, products, and transition states are calculated at the bhandhlyp/6-31g(d) level, and the vibration frequencies are optimized. According to the above four reaction paths, the transition states of each path are determined, and the geometrical structures of reactants, products, and transition states are calculated at the bhandhlyp/6-31g(d) level, and the vibration frequencies are optimized. The structure of transition state was calculated by IRC, and the corresponding reactants and structures were obtained. Because the calculation step of IRC is fixed, the structure of “reactant” and “product” found in the strict sense is not the real reactant and product, but the closest to its structure. Therefore, the optimization calculation of the two structures is carried out at the same level.
By further calculation and analysis, the structures of transition states (TS) and initial reaction products (P) corresponding to Structures I and III are selected, excluding the similar and impossible structures in the calculation. Figure 6 shows the structure of the transition states (TS) and the initial reaction products (P). From Structure I of Figure 6(a) and (b), it can be seen that it first transfers on O30 in RDX from the nitro group to the Al1 atom in the Al cluster; with the further separation of O30, it also interacted with Al11 until it completely separated from the N26 atom. From Structure III of figures, it can be seen that it first transfers on O32 in RDX from the nitro group to Al1 atom in the Al cluster. As the reaction proceeds, O32 completely transfers from N27 atom to Al8 atom.
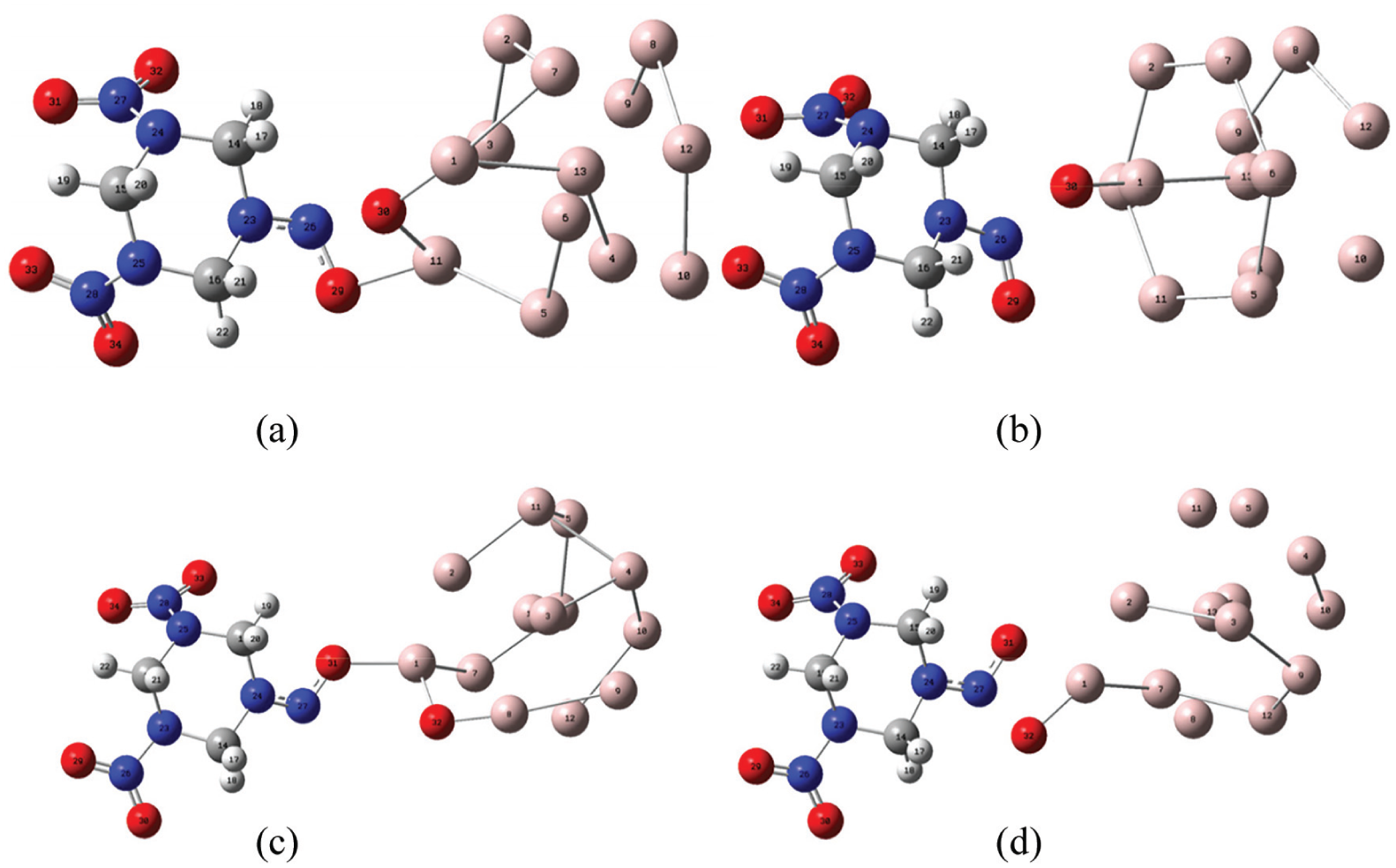
TS and product structures corresponding to Structures I and III: (a) TS of Structure I, (b) product structure of Structure I, (c) TS of Structure III, and (d) product structure of Structure III.
By calculating the energy difference between the transition state and the reactant in the structure, the activation energy required for the first step reaction is obtained. The results are shown in Table 2. From the data in the table, it can be seen that the activation energy of Structure I is 56.448 × 103 J mol−1, while that of Structure III is 93.993 × 103 J mol−1. This indicates that in RDX and Al systems, the O30 atom of RDX is more easily separated from the reaction. In addition, compared with Al11, Al1 in aluminum clusters is more vulnerable to “attack” by O atoms, which is determined by its spatial location characteristics.
Energy and activation energy of transition states (TS), reactants (R), products (P) corresponding to Structures I and III.
Reaction mechanism of AP and Al
Analysis and exploration of computing procedures
Eight structures of AP and Al were calculated according to the calculation process of RDX reaction path. The results are inconsistent with the initial hypothesis. In Structure I, for example, it is assumed that O21 first acts with Al clusters. After prolonging the bond length and fixing optimization, it is found that not only O21, but also the other oxygen atoms (O22 and O23) interact with the surface of Al clusters, but, the latter two oxygen atoms have not completely broken off from Cl atoms. The results of subsequent transition state optimization show that the transition state structure of O21 and O23 forms bonds with Al, but the only virtual frequency result corresponds to the rotation of some Al atoms in the Al cluster, rather than the stretching vibration of O21–Cl19 bond, which contradicts the original assumption that was the fracture of O21–Cl19 bond, so the “transition state structure” found is unreasonable. For the other configurations, either there is no imaginary frequency or the corresponding vibration mode is incorrect, and the appropriate transition state structure has not been found after eliminating the imaginary frequency. The main reasons for the above situation and the solutions are summarized as follows:
In searching for the transition state of the decomposition reaction, the old bonds are not always broken first, but may be broken only after the newly formed bonds are formed. In view of this, it is necessary to scan the “broken bond” and “new bond” separately until possible transition state structures are found or such scanning settings are excluded.
The steps in the chemical reaction process are not always carried out in strict sequence, and two-step reactions may occur at the same time. Therefore, if the possible structures are not found after scanning the “broken” and “new” keys, respectively, it may be necessary to scan the above two keys at the same time.
Even if the results of the previous steps are “reasonable,” there may be a mistake in the transition state in the end. Therefore, in order to ensure the accuracy, IRC analysis should be carried out after the “transition state” structure is found, and the corresponding reactant and product structure should be optimized to establish the relationship between reactant, transition state, and product.
Determine the calculation method
Based on the above analysis, the SCAN method is used to scan the reaction bond length. After optimizing the initial structure, the bond length will be lengthened or shortened gradually during the scanning process. After the scanning, the energy trends of the structures with different bond lengths will be analyzed, and the structure corresponding to the highest energy point (i.e. the most unstable structure) will be selected as the initial structure to optimize the transition state.
By further calculation and analysis, similar repetitive structures are excluded. Transition states (TS) and initial reaction products (P) corresponding to Structures III, IV, V, VI, and VIII are selected. Their structures are shown in Figures 7 and 8. From Structure I, it can be seen that O21 in AP first breaks off from perchlorate and interacts with Al1 atom in Al cluster; with further separation of O21, it also interacts with Al11 until it completely separates from Cl19 atom. At the same time, the O22 atoms also interact with the Al atoms in the Al clusters. The action process of Structure II is similar to that of Structure I. From Structure V, it can be seen that O23 in AP reacts with Al1 atom in Al cluster, and with the reaction proceeding, O23 will not completely separate from Cl19 atom. From Structure VI, it can be seen that O23 in AP first acts with Al11 atom in Al cluster. With further separation of O23, O23 and H17 atoms form hydroxyl radicals completely separated from AP molecule. At the same time, O21 and O20 also interact with Al4 atom in Al cluster. The interaction process of Structure VIII is similar to that of Structure V, and that of Structure VII is similar to that of Structure VI.
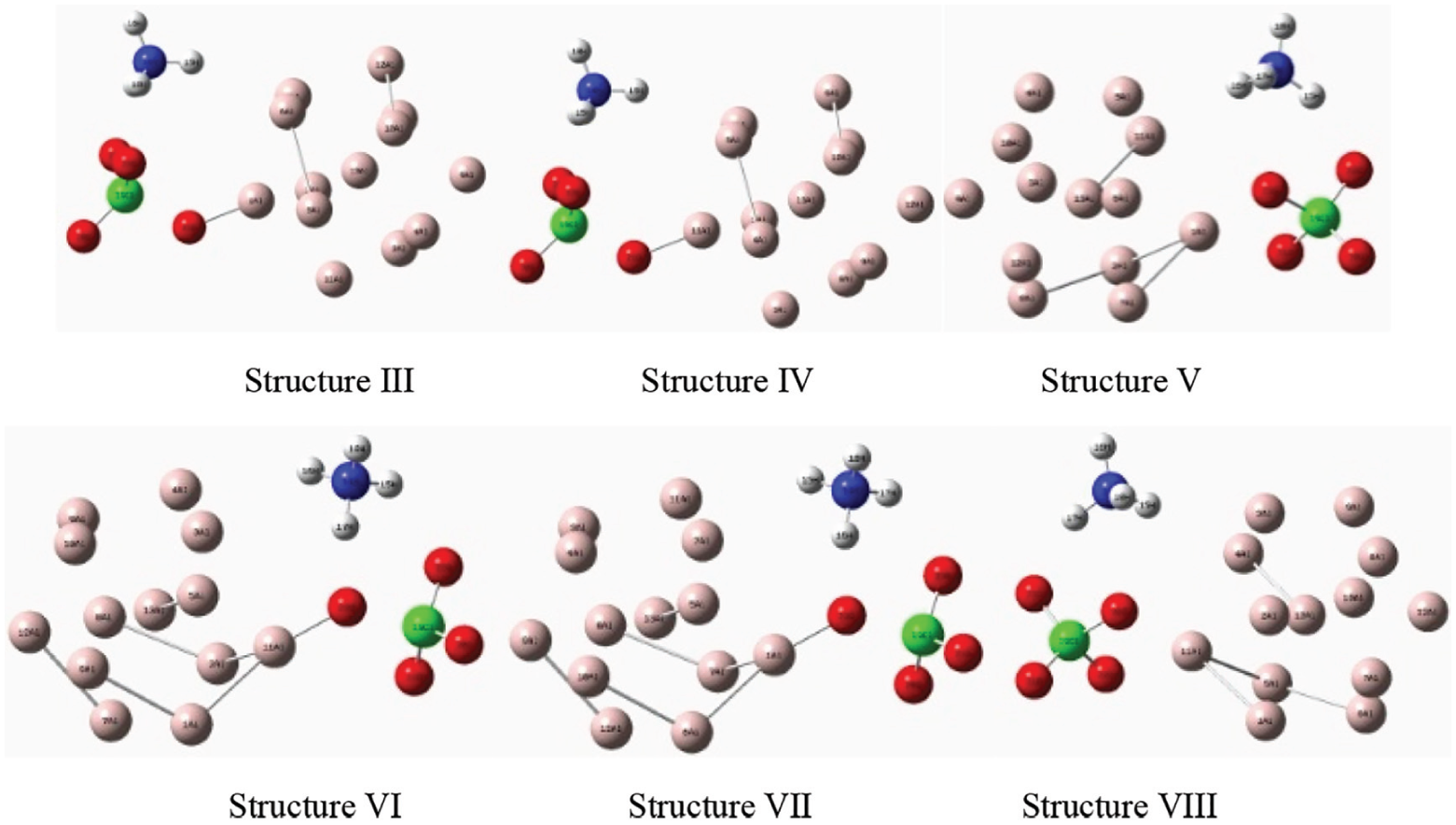
Transition states (TS).
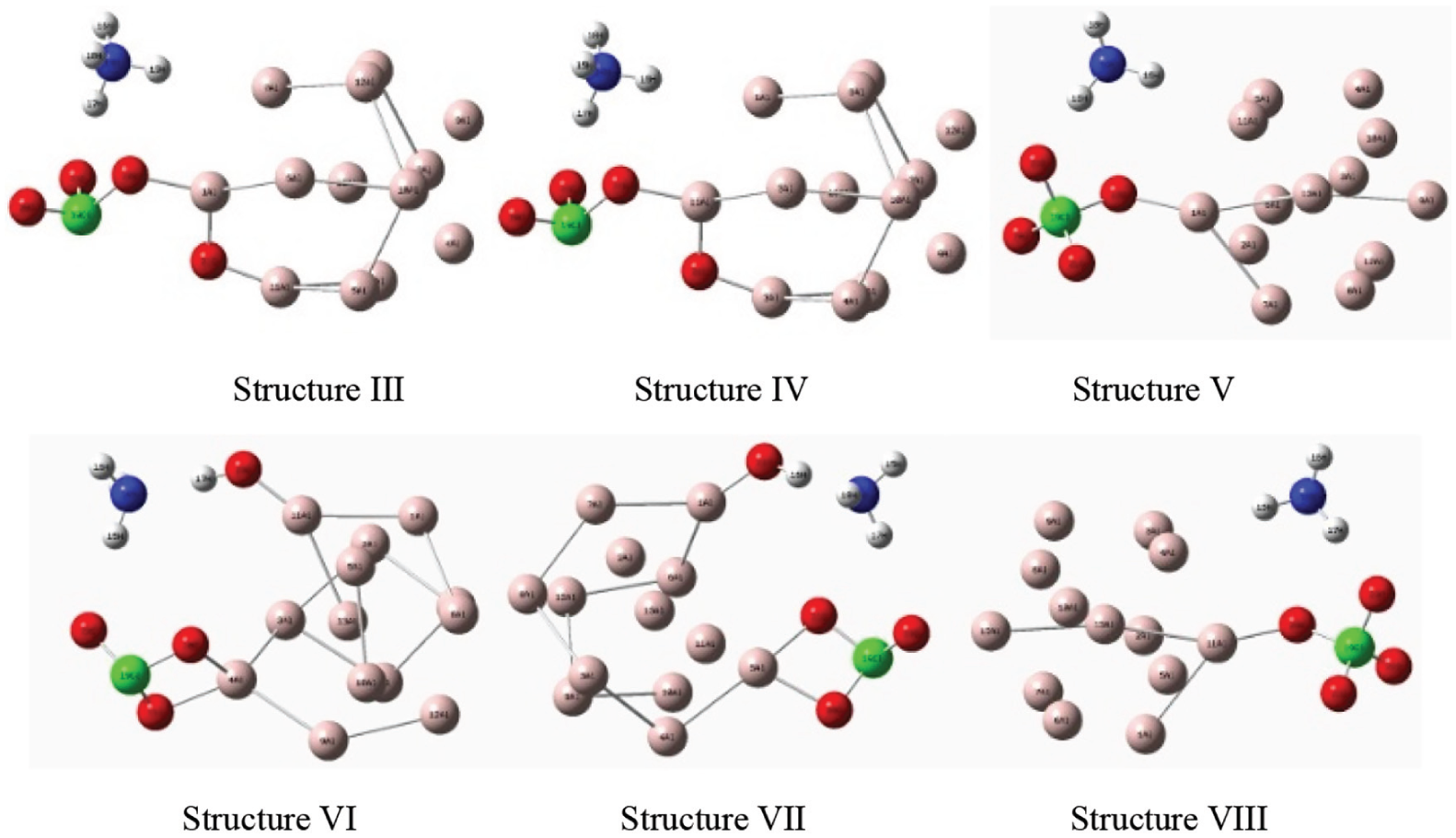
Reaction products (P).
The SCAN scanning and transition state analysis of Al + AP structures were carried out. The repetitive and similar structures were excluded. The energy difference between the transition states and reactants corresponding to the selected structures was calculated. The activation energy required for the initial reaction was obtained. The results are shown in Table 3.
The energies and activation energies of transition states (TS), reactants (R) corresponding to six selected structures.
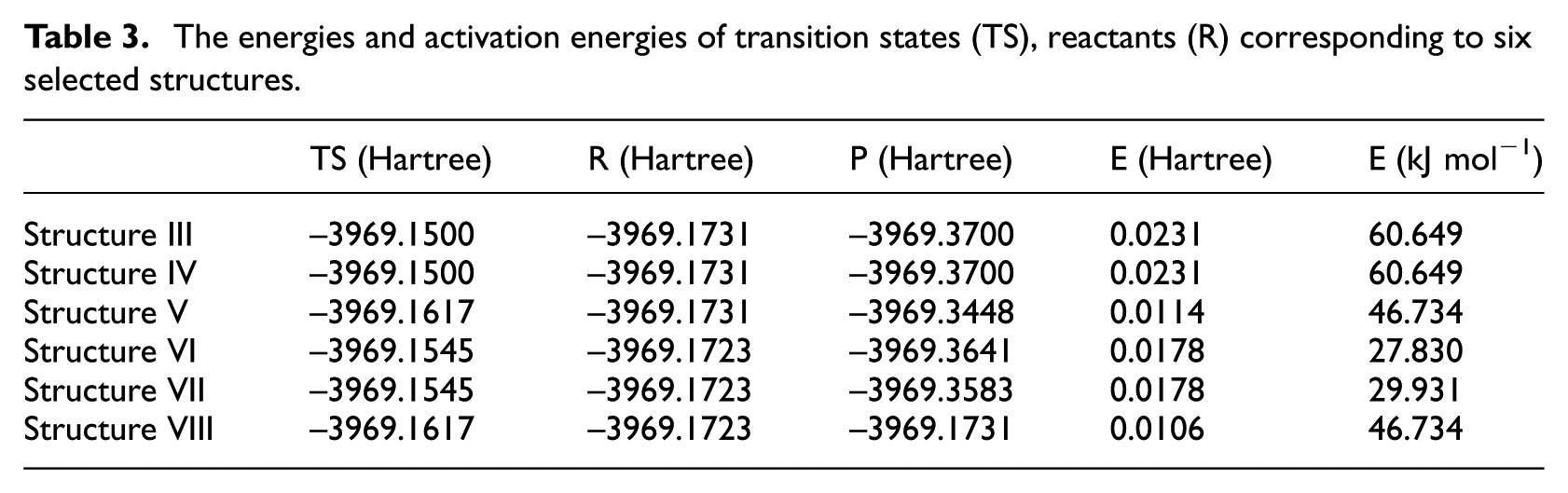
The activation energies required for Structures V and VIII (<30 × 103 J mol−1) are significantly lower than those for other reactions (>45 × 103 J mol−1), while the activation energies required for Structures III and IV are the largest (60.649 × 103 J mol−1). This indicates that in AP and Al systems, the O22 atoms of AP are more likely to break away from Cl19 and react with Al atoms. Compared with Structures V and VI, O and Al11 are more likely to react, whereas Structures VII and VIII more likely to react, that is, O and Al1 are more likely to react. Compared with Structures VI and VIII, it can be seen that the electrostatic dominant interaction has different effects on the activation energy of the reaction due to the different relative positions of ammonium ions and oxygen atoms. The interaction in Structure VI is weaker and its activation energy is lower.
Conclusion
By calculating the four Al + RDX structural models, it is known that the O30 atom of RDX first breaks off from the nitro group and is easier to break away from RDX and interact with the vertex atom of Al13 cluster. With the further separation of O30, it also acts with Al11 until it completely breaks away from N26 atom. The activation energy of this reaction is 56.448 × 103 J mol−1.
By looking for eight transition states corresponding to the initial model of Al + AP, it is known that the O22 atoms in AP are more likely to interact with the Al1 atoms in Al clusters. With the reaction proceeding, the O22 atoms will not be completely separated from Cl19 atoms, and the corresponding activation energy is 27.830 × 103 J mol−1.
It is further verified that Al powder of analytic propellant participates in the thermal decomposition reaction of melted RDX and AP in TG test and promotes the thermal decomposition reaction of both.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.