
Abstract
The density functional theory method is employed to systematically explore the mechanism of syngas methanation on the Ni4/MCM-41 catalyst surface. The calculation results show that the optimal pathway of CH4 formation is CO + H → CHO + H → CH2O + H → CH3O → CH3 + H → CH4 with the rate-determining step of CH3O direct dissociation. Because the activation energy for the direct dissociation of CH3O species is much lower than that for the CH3OH formation (198.6 vs 264.8 kJ mol−1), there is almost no by-product CH3OH that appeared in the products of the syngas methanation over the Ni4/MCM-41 catalyst. Compared with other conventional nickel-based methanation catalysts, Ni4/MCM-41 catalyst is an excellent methanation catalyst with high selectivity of CH4.
Introduction
As one of the fossil fuels, natural gas is considered to be an efficient and clean energy source. In recent years, due to the lack of reserves, the rising price, and the increasing consumption of natural gas, coal to synthetic natural gas (SNG) technology has attracted more and more attention. 1 Syngas (H2/CO) methanation is a key reaction in coal to SNG technology. Many fields, such as ammonia synthesis reaction, fuel cell, and so on, also involve CO methanation reaction, which is used to remove the trace CO. 2 The research and development of high efficiency catalyst for syngas methanation is one of the key points in coal to SNG technology. Due to the low cost, high activity, and good selectivity, nickel-based catalysts are the most widely used catalysts for syngas methanation. 3 The properties of supports has an important influence on the performance of nickel-based catalysts.4,5 However, syngas methanation is a strong exothermic reaction, and the active particles of the traditional catalyst are easy to sinter and deactivate due to the rapid release of reaction heat. 6
In our previous work, Ni nanoparticles supported on MCM-41 (Ni/MCM-41) catalysts showed excellent sintering resistance in syngas methanation reaction. 7 Compared with the conventional Ni/Al2O3 catalyst, Ni/MCM-41 based on the MCM-41 support exhibited the excellent catalytic activity and stability in the reaction of syngas methanation. 8 Mesoporous molecular sieve MCM-41 is considered to be an excellent support material to replace the conventional catalyst support for the strong exothermic reactions of syngas methanation, 9 methane reforming, and Fischer–Tropsch synthesis, which is due to its excellent thermal stability, chemical inertia, and high thermal conductivity.
The study of reaction mechanism is very important for the research and development of catalysts. Many studies have focused on the mechanism of CO methanation, and the most representative mechanisms include carbide mechanism and H-assisted CO dissociation mechanism.10–12 Previous studies have shown that the mechanism of CO dissociation must include H-assisted CO dissociation in addition to direct CO dissociation due to the high activation energy of CO dissociation. 13 However, continuous hydrogenation of CHxO or CHxOH intermediates in CO methanation may produce by-product of CH3OH. 14 Wang et al. 15 reported that the breakage of C–O bond of CHxO (H) intermediate and the decrease of CH3OH by-product were beneficial to the increase of CH4 yield on the surface of Ni4/γ–Al2O3 catalyst. Ghanashyam and Asoke 16 found that the adsorption or binding energy of CO on Ni4 cluster is a thermodynamically feasible process at normal condition whereas dissociation is not feasible.
To date, although much work has been done to study the CH4 formation from CO over nickel-based catalysts, it is necessary to investigate the mechanism of syngas methanation on the Ni4/MCM-41 catalysts. Density functional theory (DFT) is of great significance in the study of catalyst performance, catalyst screening and design, and investigation of the reaction mechanism. 17 Hence, it is important to study the reaction mechanism of syngas methanation based on Ni4/MCM-41 catalyst at atomic and molecular levels. The mechanism of syngas methanation on Ni4/MCM-41 catalyst surface is systematically studied by DFT method in this article.
Computational models and methodology
Computational models
We set the parameters of MCM-41 model according to the literature. 18 The two walls of MCM-41 are connected by bridging the oxygen center (Ob, Figure 1(a)). From the structural optimization of the flat plate model, 535 pm for the edges of a hexagonal unit cell were obtained and α was 60°, and the optimized structural parameters were 165 pm for Si–Os, 167 pm for Si–Ob, and Si–Ob–Si = 180°, in which, Os denotes a surface O center, Figure 1(a). MCM-type single-wall structure has recently been employed to model a silicate overlayer on a molybdenum support. 19 For the adsorption on MCM-41 carrier, a (2 × 2) unit cell of the ideal substrate (Figure 1(a)) was employed and a defect site as Ob vacancy was constructed, in which the Si center of the “bottom” layer is saturated with the silanol group. The unconstrained optimization was performed for the structures of the MCM-41 model. The slabs were separated by setting a vacuum region of 15 Å in the z direction, which were perpendicular to the surface.
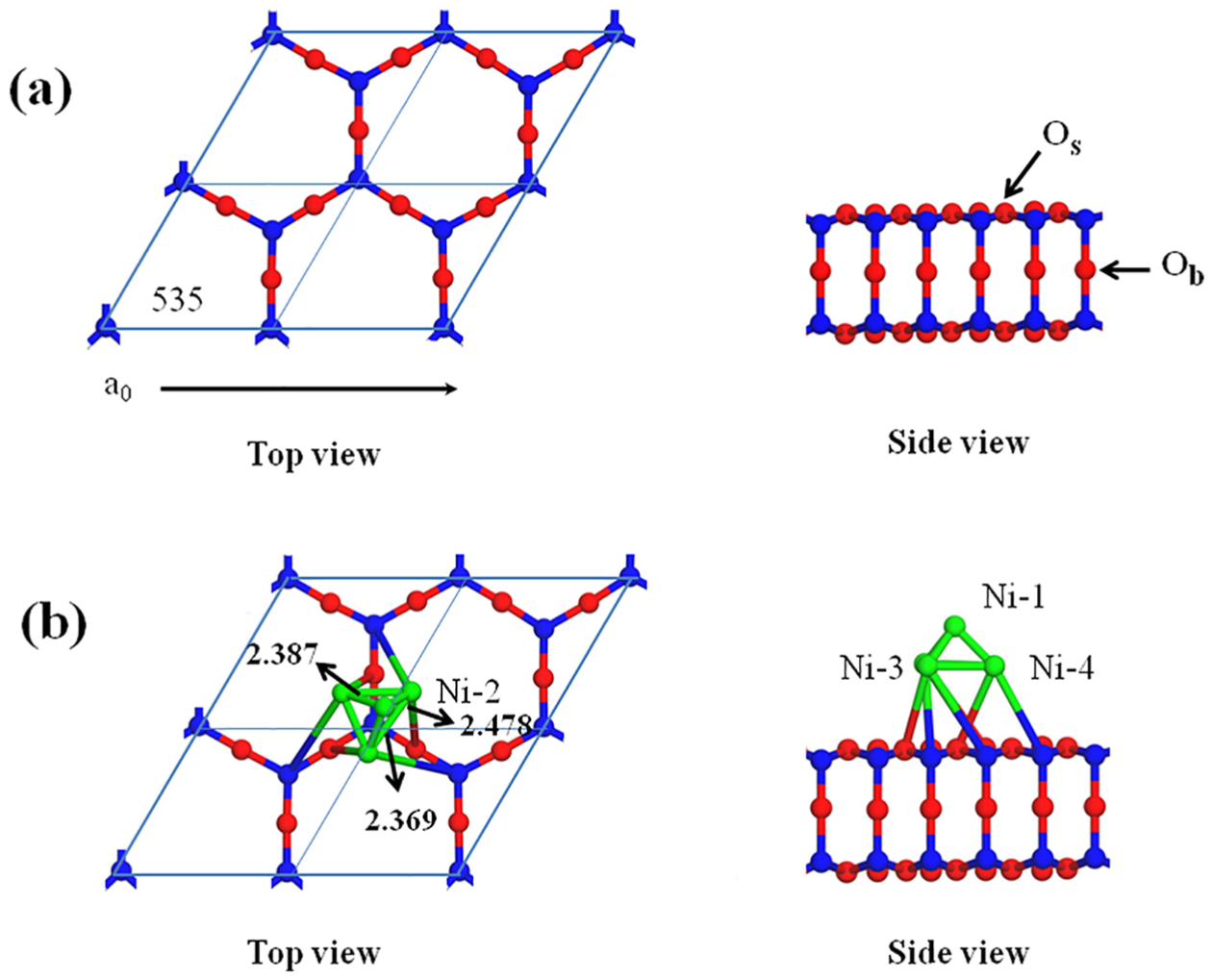
The top and side views of (a) the MCM-41 surface and (b) Ni4 cluster supported on surface. Blue balls stand for Si atoms, red balls stand for O atoms, and green balls stand for Ni atoms.
In addition, the Ni4 cluster, which is the smallest three-dimensional structure of nickel atoms, is chosen to study the interaction between nickel nanoparticles and between nickel particles and support. 20
Computational methodology
Dmol3 code on Materials Studio 7.0 Package was employed to carry out DFT calculations. 21 The correction and exchange effects were studied with the Perdew–Burke–Ernzerh (PBE) and generalized gradient approximation (GGA) functional. An effective core potential (ECP) was adopted to replace the inner electrons of the nickel atoms based on the double numerical plus polarization (DNP) basis sets, which were kept frozen. 22 In adsorption studies, a grid of (5 × 5 × 1) k-points was employed for the surface model of the mesoporous molecular sieves MCM-41. A 10 × 10 × 10 Å 3 unit cell was adopted to calculate the free adsorbates, and an all-electron basis set was used to calculate other atoms. The convergence criterion for the energy was set at 2.0 × 10−5 Hartree with the spin unrestricted condition. The convergence criterion for maximum displacement changes and maximum force were set at 5.0 × 10−3 Å and 4.0 × 10−3 Hartree Å−1, respectively. The thermal smearing value and the k-point was set as 0.005 Ha and 4 × 4 × 1, respectively. The self-consistent field (SCF) convergence was 1 × 10−5 Hartree. The reaction transition state was identified by the linear synchronous transit (LST/QST) of the Dmol3 code. 23 Adsorption energy is defined as follows
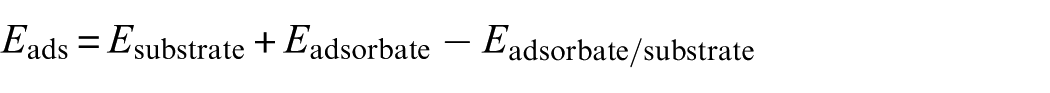
in which Eadsorbate/substrate represents the total energy of the adsorbate–substrate system in equilibrium; Esubstrate represents the total energies of the substrate; and Eadsorbate represents the total energies of the substrate, the free adsorbate.
Results and discussion
Ni4 cluster supported on the MCM-41 surface
Figure 1 shows the configurations of MCM-41 and Ni4 cluster supported on MCM-41 surface. Because the tetrahedral structure is more energy-efficient among the three possible structures of Ni4 cluster (linear, rhombic, and tetrahedral), we chose the three-dimensional Ni4 with tetrahedral structure to adsorb on the surface of MCM-41 as the catalyst to study the mechanism of CO methanation. Figure 1(b) shows the most stable adsorption configuration of the Ni4 cluster on MCM-41 surface and the key structural parameters. The relative bonding energy of Ni4 cluster on MCM-41 surface is higher than that of Ni4 cluster on Al2O3 surface (67 vs 479 kJ mol−1), which may effectively inhibit the reunion of Ni atoms on the surface of MCM-41 in the process of CO methanation.
Adsorption of species on Ni4/MCM-41 surface
There is no doubt that the adsorption of species plays an important role in the catalytic process of CO methanation. Therefore, the species adsorbed at different sites on the surface of Ni4/MCM-41 catalyst involved in CO hydrogenation reaction are studied in this section, and Figure 2 shows the calculated adsorption energy and preferred adsorption sites of adsorption species involved in the CO hydrogenation reaction on the Ni4/MCM-41 catalyst surface. Because hydrogen molecules could easily dissociate into atomic hydrogen on the surface of nickel metal12,15,24,25 any possible adsorption sites on the surface of Ni4/MCM-41 catalyst will be filled with high concentration of H atoms and highly efficient adsorption under the condition of syngas methanation. Detailed configuration descriptions of adsorbed species on the surface of Ni4/MCM-41 catalysts involved in the CO methanation are provided in the Supplementary Information.
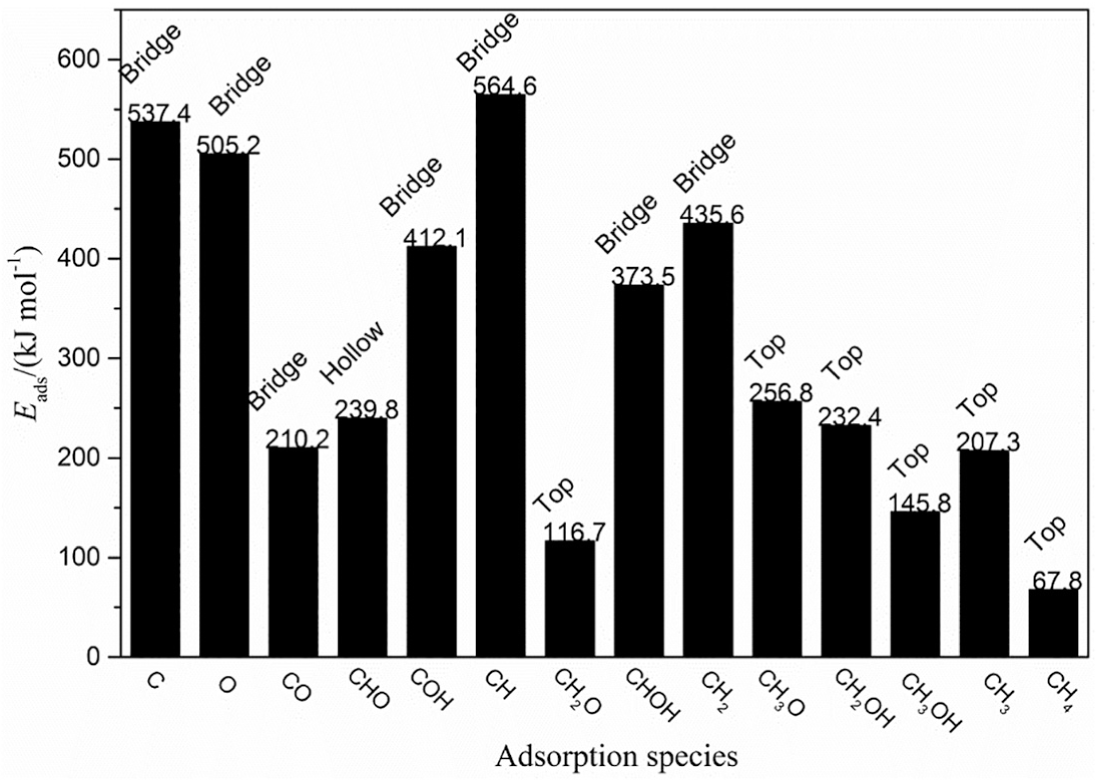
The calculated adsorption energy and preferred adsorption sites of adsorption species involved in the CO hydrogenation reaction on Ni4/MCM-41.
Reaction pathways of syngas methanation
The calculation of activation energy (Ea) is employed to investigate the reaction mechanism of syngas methanation after identifying the preferred adsorption sites of the important possible species involved in the reaction. There are a total of 12 elemental steps involved in methanation of syngas to methane, as shown in Figure 3. The calculated activation energies (Ea) and reaction enthalpies (ΔH) for each elemental steps over Ni4/MCM-41 surface are also shown in Figure 3. DFT calculation is adopted to evaluate multiple reaction pathways to obtain the optimal pathways on the Ni4/MCM-41 catalyst.
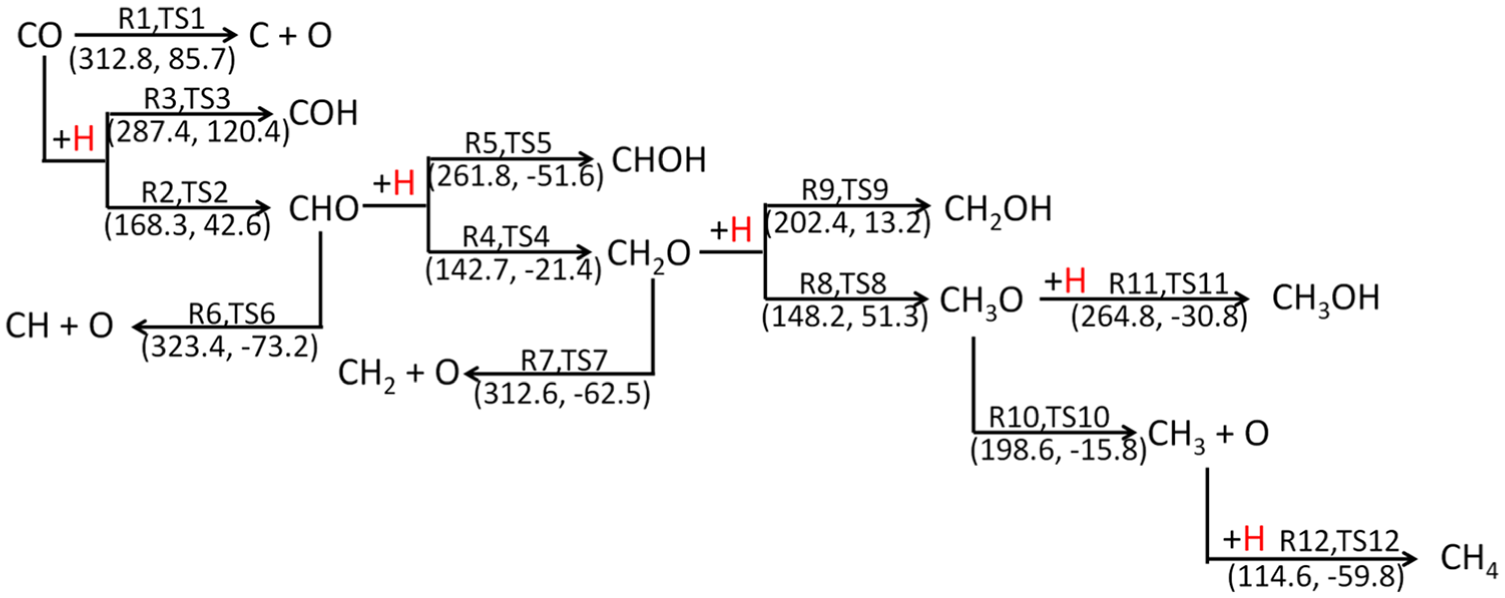
Elementary steps and the corresponding calculated Ea and ΔH values involved in CO hydrogenation over Ni4/MCM-41 surface. TS denotes the transition state, the first number in parentheses denotes the activation energy (Ea), and the second number denotes the reaction enthalpy (ΔH) with the unit of kJ mol−1.
Formation of formyl (CHO)
As shown in Figure 3, two kinds of reactions may occur on the CO species adsorbed on the Ni4/MCM-41 surface, including the adsorbed CO species directly breaking into C and O atoms and reacting with H to form formyl species (COH or CHO) by hydrogenation. The relative energy files of the three possible reactions (R1–R3) and the structures of the initial states (ISs), transition states (TSs), and final states (FSs) are shown in Figure 4. It can be seen that the activation energies for direct dissociation of CO, CHO, and COH species formation are 312.8, 168.3, and 287.4 kJ mol−1. Compared with the direct dissociation of CO, the activation energy required for the formation of CHO and COH is lower, with the CHO species as the lowest. These results indicate that the first intermediate of CO hydrogenation reaction should be CHO species based on activation energy.
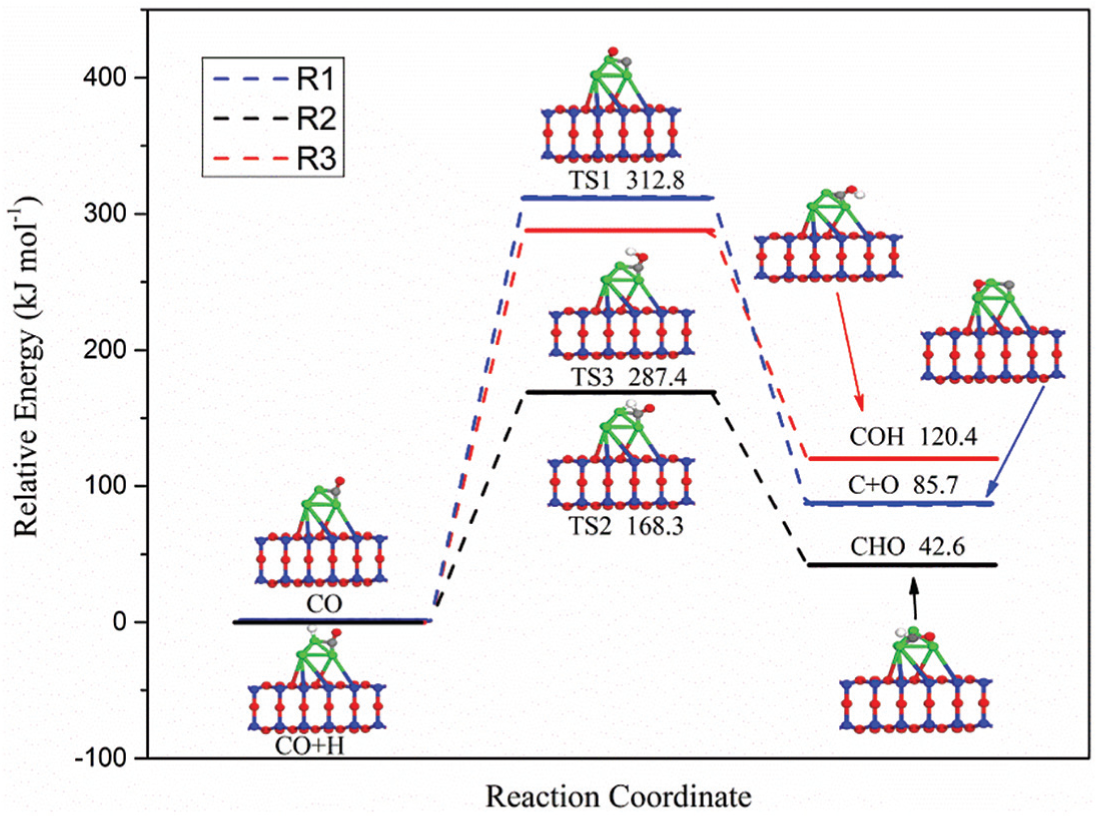
Potential energy profile for R1–R3 over Ni4/MCM-41 catalyst with the configurations of initial states, transition states, and final states.
During the formation of CHO species, CO and H coadsorbed on the surface of Ni4/MCM-41 catalyst, where CO is strongly present at the bridge site through C atom with the C–Ni–1 and C–Ni–2, and the H is located at the top of the Ni4 atom with an H–Ni–1 bond. The formation of TS2 is through the bonding of H atom with C atom of the absorbed CO, which is an endothermic step with a reaction enthalpy of 42.6 kJ mol−1. In TS2, initially formed CHO species are bound to the substrate by the C–Ni–1, C–Ni–2, and H–Ni–1 bonds. The lengths of H–Ni–1, C–O bonds, and newly formed C–H bonds are 1.725, 1.328, and 1.346 Å, respectively. Subsequently, TS2 transforms into a more stable form, the final CHO species.
Formation of formaldehyde (CH2O)
The CHO intermediate formed in R2 may dissociate directly into CH and O atom or hydrogenates with other H atom to form hydroxymethylene (CHOH) or formaldehyde (CH2O) intermediates. The three element steps (R4–R6) corresponding to the above two possibilities are shown in Figure 3 and the corresponding configurations of ISs, TSs, and FSs and potential energy profile are presented in Figure 5. It can be seen that the activation energies for direct dissociation of CHO, CH2O, and CHOH species formation are 323.4, 142.7, and 261.8 kJ mol−1. The activation energy of CH2O formation is 180.7 and 119.1 kJ mol−1 lower than those of direct dissociation of CHO and CHOH species formation, respectively. Therefore, CHO species are more likely to form CH2O intermediate species by hydrogenation with other H atoms and continue the following reactions.
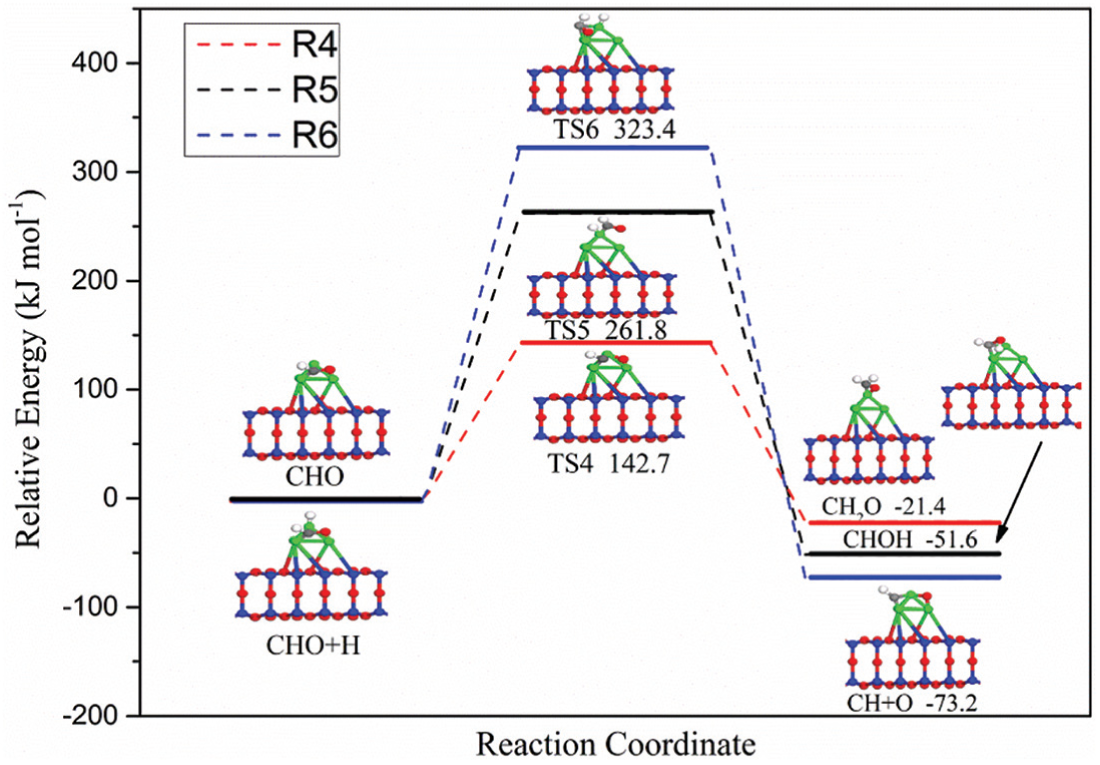
Potential energy profile for R4–R6 over Ni4/MCM-41 catalyst with the configurations of initial states, transition states, and final states.
In the pathway of CH2O species formation, CHO species and H atom are coadsorbed on the Ni4 clusters, in which CHO species occupy the hollow site of Ni4 clusters through the C and O atoms, while the H atom is located at the Ni4 clusters’ top site via the newly formed H–Ni–1 bond. Subsequently, with the breaking of C–Ni–3 bond and O–Ni–4 bond, CHO species migrate to the top site of the Ni4 cluster, forming the transition state TS5. Ultimately, the transition state TS5 is transformed into a more stable form, the final CH2O species.
Formation of methoxy (CH3O)
As shown in Figure 3, methoxy (CH3O, R8) or hydroxymethyl (CH2OH, R9) species can be formed by hydrogenation of the CH2O intermediates with an H atom. Moreover, the CH2O intermediates may also dissociate directly to form CH2 and O atoms (R7). The configurations of ISs, TSs, and FSs corresponding to the three element steps (R7–R9) and potential energy profile are presented in Figure 6. The activation energies of CH2O direct dissociation, formation of CH3O, and formation of CH2OH are 312.6, 148.2, and 202.4 kJ mol−1, respectively. Obviously, in terms of energy barrier, CH2O is more likely to generate CH3O species by hydrogenation. Therefore, CH3O species should be the third intermediate to continue the reaction.
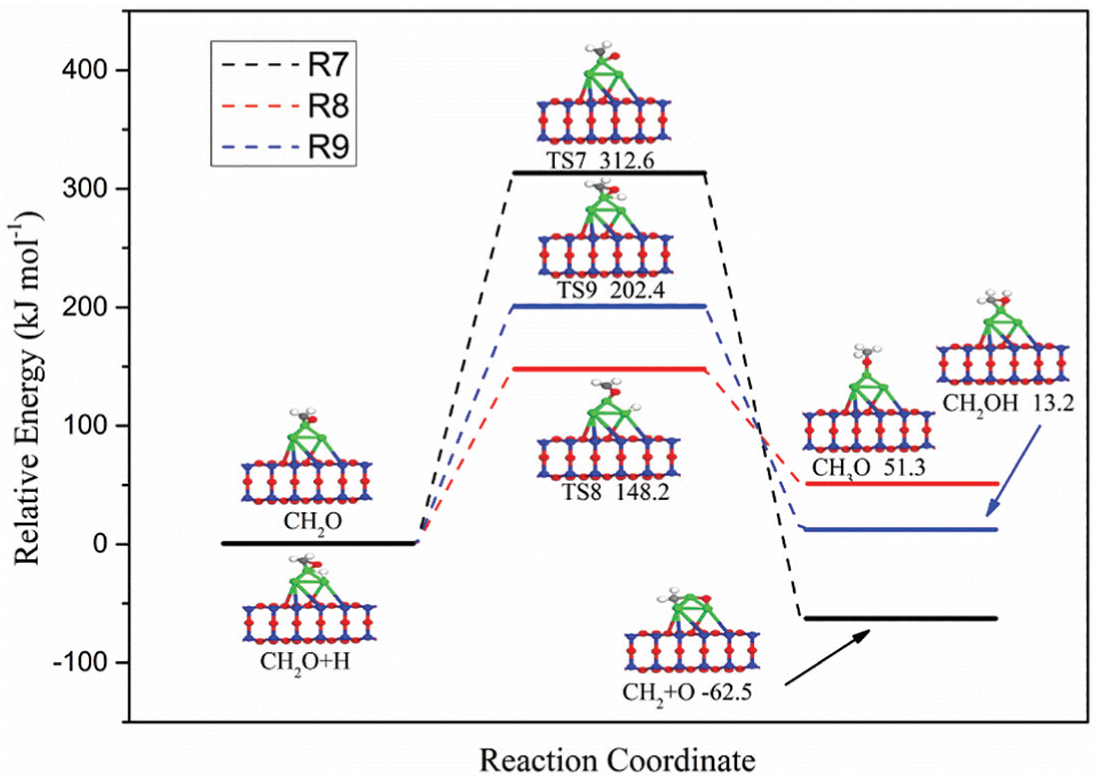
Potential energy profile for R7–R9 over Ni4/MCM-41 catalyst with the configurations of initial states, transition states, and final states.
The pathway of CH3O species formation starts from the coadsorption of the CH2O species and H atoms on the Ni4/MCM-41 surface. Initially, CH2O species occupy the top site of the Ni4 cluster via the C–Ni–1 bond and O–Ni–1 bond and the H atom is located at the bridge site of Ni–1–Ni–4. Subsequently, with the breaking of C–Ni–1 bond and H–Ni–4 bond, the coadsorption state of the CH2O species and H atom is converted into the transition state TS8. For TS8, the bond lengths of O–Ni–1 and H–Ni–1 are 1.628 and 1.014 Å, respectively, with the C–O–Ni angle of 106°. Subsequently, the transition state TS8 is transformed into a more stable form, the final CH3O intermediate species, which binds to the Ni4/MCM-41 via the O atom.
Formation of CH4
As shown in Figure 3, the preceding intermediate species of CH3O can react with an H atom to form CH3OH or dissociate directly into the CH3 species and O atoms, which could further react with one more adsorbed H atom to form the final product CH4 via TS12. The configurations of ISs, TSs, and FSs corresponding to the involved element steps (R10–R12) and potential energy profile are presented in Figure 7. It can be found that the direct dissociation of CH3O is more likely to occur than the formation of CH3OH in terms of activation energy (198.6 vs 264.8 kJ mol−1), which is consistent with the findings of Wang et al. 15 and Han et al. 25 However, unlike the results of Wang et al. 15 and Han et al., 25 the activation energy required for the CH3OH formation is 33% higher than that for the direct dissociation of CH3O. In case the CH3 intermediate species appeared on the surface of the Ni4/MCM-41 catalyst, it can react rapidly with the adsorbed H atom to form CH4, which is an exothermic reaction with 59.8 kJ mol−1.
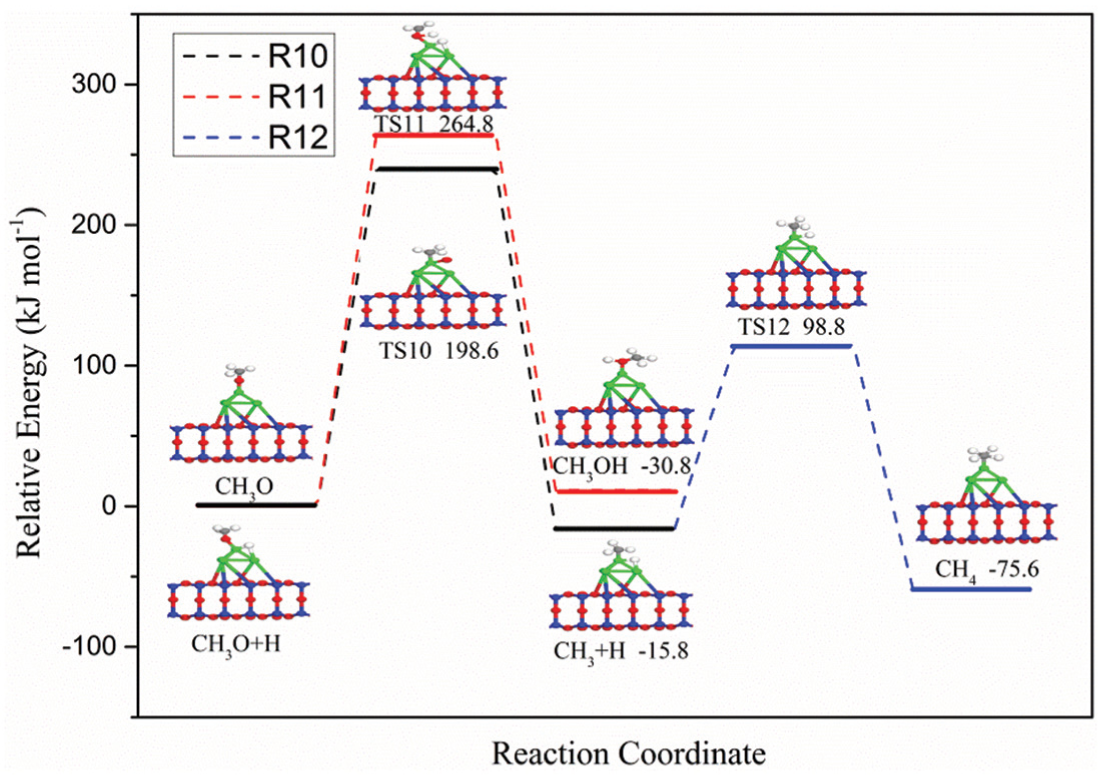
Potential energy profile for R10–R12 over Ni4/MCM-41 catalyst with the configurations of initial states, transition states, and final states.
Since the activation energy required for the CH3OH formation is much larger than that for the CH3O direct dissociation, it is almost impossible to form CH3OH by CO hydrogenation over Ni4/MCM-41 catalyst, which is consistent with our previous experimental results. 26 Therefore, the formation of CH3OH is not discussed too much. The adsorbed CH3O species are transformed into the transitional state TS10 via breaking C–O bond and the newly formed C–Ni–1 bond. Subsequently, TS10 splits into the adsorbed CH3 species and O atoms. For TS10, the CH3 is located at the top site of Ni4 cluster via the newly formed C–Ni–1 bond with a bond length of 2.024 Å. The pathway of CH4 species formation starts from the coadsorption of the CH3 species and H atoms on the Ni4/MCM-41 surface. Initially, CH3 species occupy the top site of the Ni4 cluster via the C–Ni–1 bond and the H atom is located at the bridge site of Ni–1–Ni–4. Subsequently, the H atom migrates to the C atom to form the CH4 species with the breaking of the H–Ni–4 bond via the transition state TS12.
General discussion
Based on the above-mentioned results, the optimal pathway of CH4 formation over Ni4/MCM-41 catalyst surface is CO + H → CHO + H → CH2O + H → CH3O → CH3 + H → CH4 and the rate-determining step is the direct dissociation of CH3O species, which is consistent with those on the Ni4/γ–Al2O3 15 and Ni4/SiC catalyst surfaces. 25 Figure 8 shows the potential energy profile for syngas methanation on the Ni4/MCM-41 catalyst surface. For Ni4/γ–Al2O3, 15 and Ni4/t–ZrO2 surfaces, 27 due to the approximate activation energy of CH3OH formation and direct dissociation of CH3O species, the formation of CH3OH can significantly reduce the yield and selectivity of CH4. However, according to our calculations, the activation energy required for the CH3OH formation is 33% higher than that for the direct dissociation of CH3O (198.6 vs 264.8 kJ mol−1), which may predicate that the formation of CH3OH may not occur.
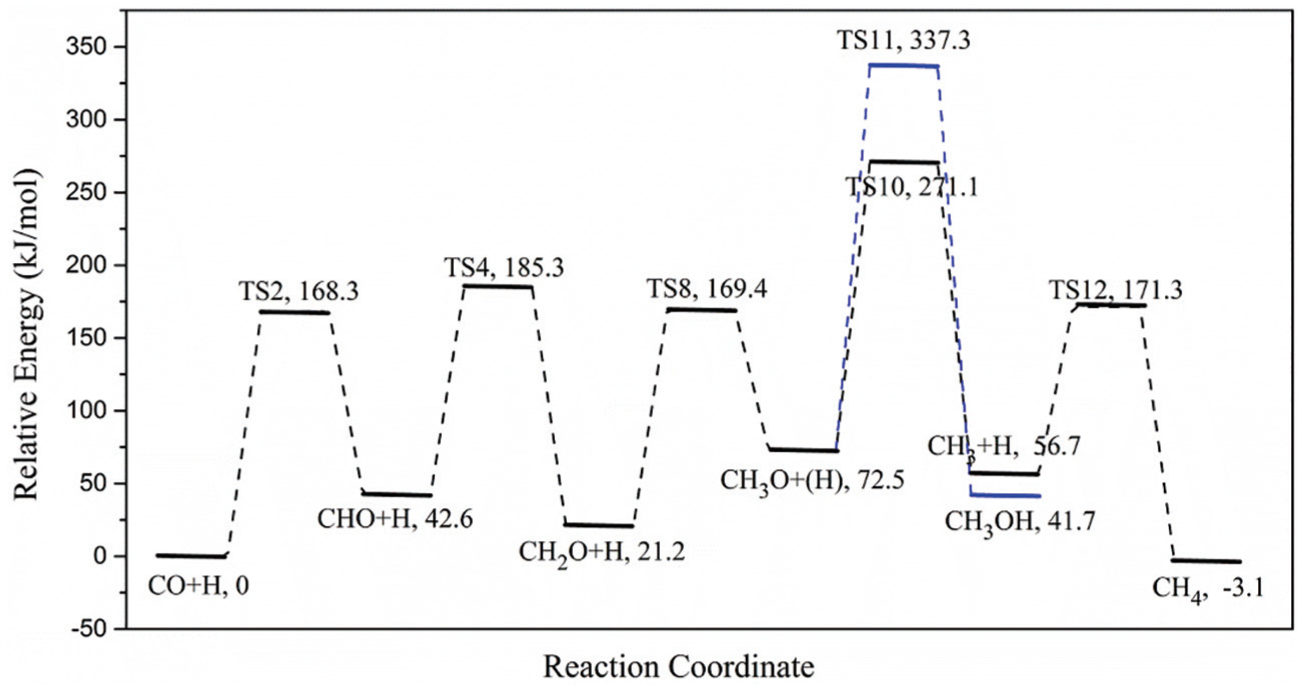
Potential energy profile for syngas methanation on the Ni4/MCM-41 catalyst surface.
As the by-product CH3OH can obviously affect the selectivity and yield of target product CH4, the calculation results in this article indicate that compared with other conventional nickel-based catalysts, Ni4/MCM-41 catalyst can significantly improve the selectivity and yield of CH4 in methanation of syngas. The calculated results in this article are consistent with our previous experimental results. 26 No CH3OH by-products were found in the products of syngas methanation based on Ni4/MCM-41 catalyst. The stability tests in our previous work show that the selectivity and yield of CH4 over Ni4/MCM-41 are much higher than those over Ni/γ–Al2O3 and Ni/SiO2 in the syngas methanation reaction, which verifies the validity of our calculation results.
Conclusion
The reaction mechanism of syngas methanation based on Ni4/MCM-41 catalyst is systematically studied by the DFT calculation method. The whole process involves 12 element steps, and the final optimized pathway of CH4 formation is CO + H → CHO + H → CH2O + H → CH3O → CH3 + H → CH4. As the rate-determining step of CH4 formation, the activation energy for the direct dissociation of CH3O species is 33% lower than that for the CH3OH formation (198.6 vs 264.8 kJ mol−1). This implies that the probability of CH3OH formation in the syngas methanation over Ni4/MCM-41 catalyst is almost negligible, which is in agreement with the previous experimental results. Therefore, compared with other conventional nickel-based methanation catalysts, Ni4/MCM-41 catalyst is an excellent methanation catalyst with high selectivity of CH4.
Supplemental Material
Supporting_information_1 – Supplemental material for Study on syngas methanation mechanism over Ni4/MCM-41 catalyst based on the density functional theory
Supplemental material, Supporting_information_1 for Study on syngas methanation mechanism over Ni4/MCM-41 catalyst based on the density functional theory by Jiaying Zhang in Progress in Reaction Kinetics and Mechanism
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors gratefully acknowledge the financial support from Doctoral Foundation of Shijiazhuang University of Applied Technology (15YB001).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.