
Abstract
Glyoxal can be important in atmospheric chemistry in terms of its ability to convert to secondary organic aerosols. In this study, the glyoxal-breaking reaction by two atmospheric active radicals, NO2 and NH2, has been investigated at the B3LYP and M06-2X levels in connection with 6-311++G(d,p) basis set. The formation of the most stable adducts from glyoxal with NO2/NH2 radical requires two hydrogen atom transfers. The accuracy of the predicted mechanisms in describing the hydrogen transfers was confirmed by atoms-in-molecules calculations and natural bond orbital analysis. The calculated results predict that hydrogen transfer process in both reactions at the M06-2X level is favourable from the kinetic and thermodynamic points of view. In the natural bond orbital analysis, the stabilization energy, E(2), delocalization corrections, at the B3LYP level is much higher than the same results at the M06-2X level (nearly twice). The activation thermodynamic parameters show that the first steps of the two reactions have lower barrier energy than the second steps. The Gibbs free energy values estimate that adducts of both the reactions at the mentioned method are spontaneous. The whole reaction of glyoxal + NH2 is more favourable than the whole reaction of glyoxal + NO2. The rate constants were calculated for the mentioned pathways using transition state theory for bimolecular steps and the fitted equations are reported.
Introduction
Glyoxal (HCO)2 is the simplest and smallest α-dicarbonyl compound. It can be produced in the atmosphere via the oxidation of precursor organic molecules originating from biogenic and anthropogenic emissions. 1 However, glyoxal can be introduced via some volatile organic compounds (VOCs) as sources of air pollution. 2 Glyoxal is considerably more volatile than most other compounds found in the particulate phase, with a vapour pressure of nearly 18 Torr at 20°C. 3 Also, it is known that the glyoxal has attracted many researchers to study its behaviour due to its great ability to act as a tracer for isoprene and other biogenic emissions. 4 However, this molecule makes a major contribution to the creation of secondary organic aerosols (SOAs) and because it has a high rate of adsorption into aerosols, it can make the SOAs larger. 5 It is notable that the formation of SOAs must be restricted due to their adverse health and climatic effects. Hence, glyoxal can be considered of great importance in atmospheric chemistry for limitation of SOA formation.3,4 The photolysis and reactions of glyoxal with some active components in the atmosphere are important to formation of undesirable products. 1 Due to the importance of glyoxal, its reactions with different atmospheric oxidants have attracted much attention of researchers. 6 Glyoxal can be broken in the atmosphere via reacting with active species such as OH, NH2, O3, NO2 and NO3. 7 NO2 and NH2 radicals are of much interest in the earth’s atmosphere. The NH2 or amidogen radical in the atmosphere has been made by photolysis of NH3. 8 Amidogen in the atmosphere is also produced by the oxidation of ammonia. 9 However, NO2 radical is a nitrogen-centred radical. It is emitted from motor vehicles in a fossil fuels burning. 10 NO2 is created by heating natural N2O4–NO2 mixtures ordinarily to such a temperature that a high concentration of NO2 species is formed. 11 Great efforts have been made to investigate the reaction of glyoxal and active species in the atmosphere experimentally. Because of the differences in experimental and atmospheric conditions, they are still under debate. 12 A good knowledge about the structure of all species involved in these reactions, for example, the charge density, nature, bond lengths, type of bonds and the various interactions of glyoxal and active components, would give a key to understanding their effective interactions. Also, it is notable that hydrogen transfer between glyoxal and reactive components is one of the most challenging issues. Theoretical calculations are suitable not only in the description of structural properties for all the cases mentioned above but also in the explanation of mechanisms of reactions and design of new pathways with desirable products from the thermodynamic and kinetic points of view. 13
To our knowledge, no calculations with the density functional theory (DFT) method and natural bond orbital (NBO) analysis have yet been performed to investigate the structural characterizations on the intra- and intermolecular interactions in glyoxal-active radical systems. Thus, in this study, the DFT method was employed to investigate the reactions of glyoxal with two active radicals, namely, NO2 and NH2, in the atmosphere. In this study, two comparisons were made: the first is a comparison of the mechanisms of reactions of NH2 and NO2 radicals with glyoxal. Both radicals have a nitrogen atom in the centre while varying in size and structure. Also, NO2 can react via all three of its atoms. The effects of these differences on the mechanisms of the reactions were studied. The second is the comparison of the two levels of DFT with different functionals and their effects on the results of this work. In the course of these comparisons, the main purpose was to investigate the effect of these two radicals on glyoxal in the atmosphere and to convert it into less harmful species. The results have allowed us, in addition to understanding that the M06-2X level is better for calculating such reactions, to study the mechanism of hydrogen transfer in these reactions.
Computational methods
The DFT is an affordable method. It costs a little time with fairly accurate results but it always has many shortcomings. The DFT computations were performed at the B3LYP/6-311++G(d,p) level as a popular method and the M06-2X/6-311++G(d,p) level as the most highly accurate functional for study of thermochemistry, kinetics and non-covalent interactions to get optimized geometries of glyoxal and the two oxidants, NO2 and NH2, and their interactions. One of the most important of these is the stretching of bonds into dissociated fragments. 14 It was proven that M06-2X covers medium-range electron correlations. 15 The M06-2X functional has double the amount of non-local exchange (2×) so it has a high non-locality functional, but, of course, it is parametrized only for non-metals. 16 In this study, the species involved in the reactions have non-covalent interactions and hydrogen bonding in some cases. However, we investigated the effects of M06-2X calculations on the results. All optimized structures were confirmed to be of minimum energy conformation except for NO2 radical, which is explained in section ‘Results and discussion’. At the optimized structures of these molecules, the frequency modes were obtained to prove that true minima were found on the potential energy surfaces. Calculations of intrinsic reaction coordinates (IRCs) were also performed at the same levels to confirm the estimated transition state (TS) geometries between reactant complexes (CR1s and CR2s) and product complexes (CP1s and CP2s) or some intermediate structures in these reactions.
Atoms-in-molecules (AIM) calculations were performed to investigate the interaction of AIM and complexes by AIM2000 software. According to AIM theory, the Laplacian of the electronic charge density
The natural bonding orbital (NBO) analysis was performed at the B3LYP and M06-2X levels of theory in order to investigate the electronic structures of the optimized geometries corresponding to charge transfer between glyoxal and other reactants. NBO calculations can examine all possible interactions between ‘filled’ Lewis-type NBOs (donor) and ‘empty’ non-Lewis-type NBOs (acceptor) and estimate their energetic importance (‘2e-stabilization’ energy) by second-order perturbation theory. These interactions are referred to as ‘delocalization’ corrections to the zeroth-order natural Lewis structure because they lead to donation of occupancy from the localized NBOs of the idealized Lewis structure into the empty non-Lewis-type orbitals and thus to departure from the idealized Lewis structure description.
For each donor NBO (σ) and acceptor NBO (σ*), 17 the stabilization energy E(2) associated with delocalization σ to σ* is estimated as

where
Results and discussion
The geometry of the stationary points of the reactions, gyloxal + NO2 (radical) and gyloxal + NH2 (radical), was completely optimized at the B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p) levels of theory. The better results at M06-2X/6-311++G(d,p) level (all of the optimized structures at this level have lower energy than the other level) led us to focus on it. In the supporting information, the obtained geometrical properties of glyoxal are compared with some results. 20 The comparisons confirm that our selected methods are sufficiently accurate. To our knowledge, other species that are reported in this work are not reported elsewhere, so comparisons are not possible. In order to study the mechanisms of all reactions, AIM and NBO analyses were carried out under similar conditions to describing the hydrogen transfers in these reactions. The formation of the most stable adducts from glyoxal with NO2/NH2 radical requires two hydrogen transfers in two steps. However, the investigation of AIM results is a challenging issue for hydrogen transfer, and this study provides a starting point to perfect modelling of the hydrogen transfer during glyoxal + NH2/NO2 radical reactions.
Gyloxal + NO2 reaction
The mechanism of the first and second hydrogen transfers and the formation of CO and NHO2 molecules as final products can be represented in two steps by the following equations
Note as mentioned earlier, NO2 radical is a nitrogen-centred radical and it has an unpaired electron on nitrogen and it can be close to glyoxal by its N atom. Of course, due to the resonance effect, there is also the probability of closing the O-head.
Also, among the NO2 conformers, the relaxed (bent) conformer with an angle of about 134.9° is the most stable. We also used another stable conformation, the triangular structure, with an angle of about 65.2° that it has 77.22 kcal mol−1 of energy more than the bent NO2, but this form can be created in the vibrationally activated level at 786.5 cm−1. Our computations showed this structure can easily be closer to glyoxal via N due to the spatial position and it gives us the opportunity to compare it with the glyoxal + NH2 radical reaction. The studies on the distribution of Mulliken charges on these two structures (bent and triangular NO2) showed that in the triangular form, N is completely negative, while in the bent form of NO2, the O atoms are the negative centres of the molecule (Figure 1), in that way, the bent conformer enters the reaction by the O-end and hence creates CR1(O), TS1(O) and INT1(O) (Figure 4).
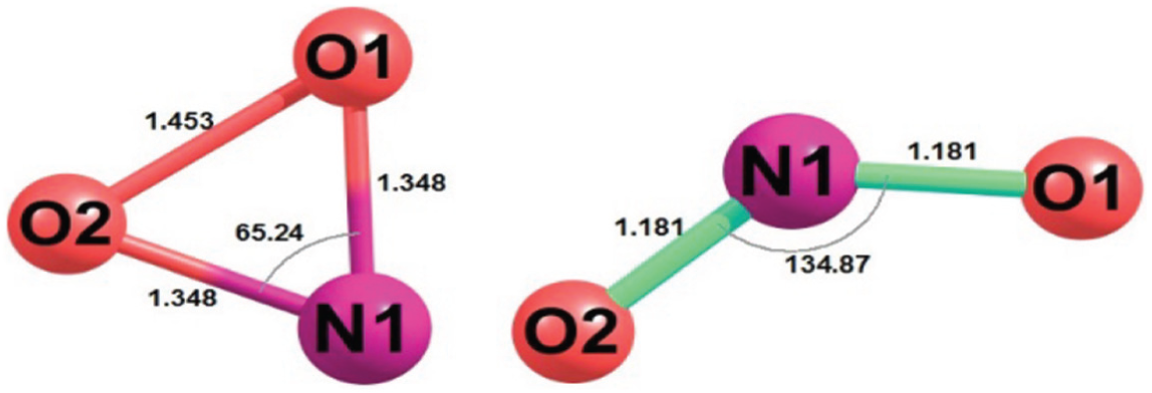
Two structures of NO2 radical: left: triangular form; right: relaxed form.
The geometries of all species involved in the two steps of reaction by the N-end of triangular NO2 contain CR1(N), TS1(N), CP1(N), CR2(N), TS2(N) and CP2(N), optimized at the M06-2X/6-311++G(d,p) level as presented in Figure 2. The pre-reactive complex formation has great importance in the processes of chemical reactions and its formation is the initial step of this reaction. The complex between glyoxal and the radical in the first step of the above reaction is labelled as CR1(N).
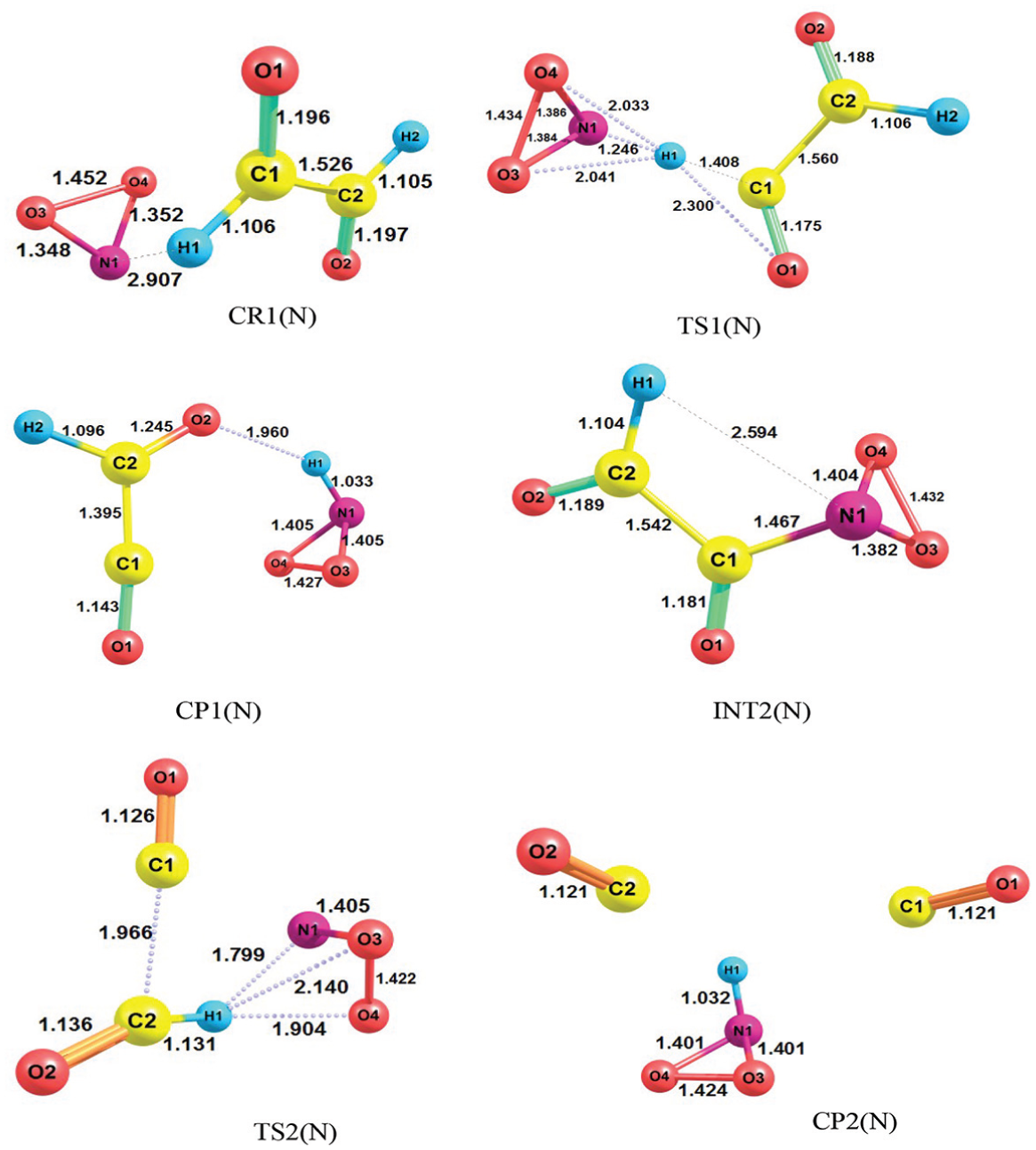
Geometries of all species involved in reactions of glyoxal + triangular form of NO2 radical containing CR1(N), TS1(N), CP1(N), INT2(N), TS2(N) and CP2(N) at the M06-2X level (bond distances are in angstrom).
CR1(N) is formed as the N atom of the triangular form NO2 approaches the hydrogen atom of glyoxal. The length of distance is 2.907 Å in the reactant complex (CR1(N)). It is notable that the length of the N–H bond calculated at M06-2X level is 0.515 Å shorter than that of the B3LYP level. A complex which is labelled as CP1(N) is produced via passing of the CR1(N) through the corresponding TS (TS1(N)). The important bond observed in the product complex is O2–H1 with the length of 1.960 Å. The NHO2 molecule and the HC2O2 radical are connected by the electrostatic interaction between the oxygen atom of the HC2O2 radical and the hydrogen atom of the NHO2 in the CP1(N) structure; this bond can be a hydrogen bond. Afterwards, the decomposition of CP1(N) and the first hydrogen transfer lead to give the NHO2 molecule and the HC2O2 radical separately as products. This step of the reaction is endothermic and non-spontaneous with almost +12.0 kcal mol−1 on the standard heat of formation and +11.7 kcal mol−1 of ΔG°. However, with certain wavelengths of sunlight, this reaction may occur in the atmosphere. During step 2, the NO2 and the HC2O2 radicals act as original reactants and form the INT2(N) structure. When the distance between C1 and N1 becomes 1.467 Å, the INT(N) will form and in this intermediate, the distance between N and H is 2.594 Å. The INT(N) is transformed to the CP2(N) after passing through the second TS (TS2(N)). The final products (2CO + NHO2) are formed by the second hydrogen transfer within CP2(N). Hydrogen transfers in the TS structures are schematized in Figure 2. In fact, with comparison of the bond lengths of CR1(N), CP1(N), INT2(N) and CP2(N) calculated at M06-2X and B3LPY levels of DFT, it can be understood that the bond lengths calculated via M06-2X level are shorter than the corresponding bonds at the B3LYP level.
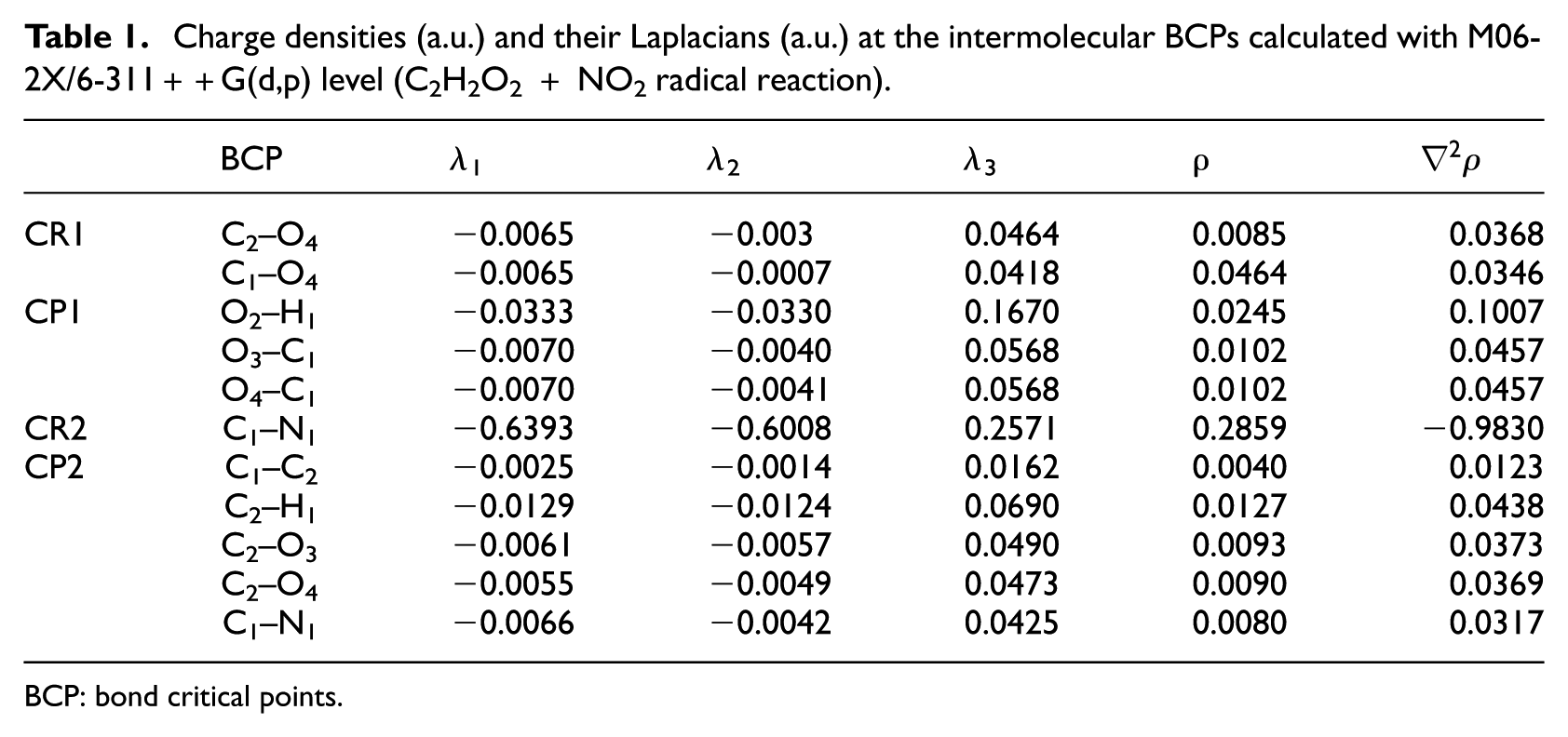
AIM analysis was done at the M06-2X level of the theory, and the results were compared with B3LYP results. Noting the topological characteristics of the BCPs during the two steps of the glyoxal + NO2 reaction at the M06-2X/6-311++G(d,p) level, the results of AIM calculation are presented in Table 1. This table tells us that the decomposition of the CP1(N) structure occurs by breaking the van der Waals bonds of O4–C1 and O4–C2 and the hydrogen bond of O2–H1. The BCPs for the certain bonds of these species are also depicted in Figure 3. As seen in this figure, the BCPs of the CR1(N) are created by the interaction between one of the oxygen atoms of NO2 and both the carbon atoms of glyoxal, thus forming a three-membered (O–C–C) ring structure in CR1(N). This complex is converted into CP1(N) through the TS1(N) structure. As shown in Figure 3, the product complex in the first stage has a six-membered ring structure that has one of its sides a hydrogen bond. This breaks to the first products, HNO2 and HC2O2, by breaking the hydrogen bond and the weak van der Waals bonds.
Charge densities (a.u.) and their Laplacians (a.u.) at the intermolecular BCPs calculated with M06-2X/6-311++G(d,p) level (C2H2O2 + NO2 radical reaction).
BCP: bond critical points.
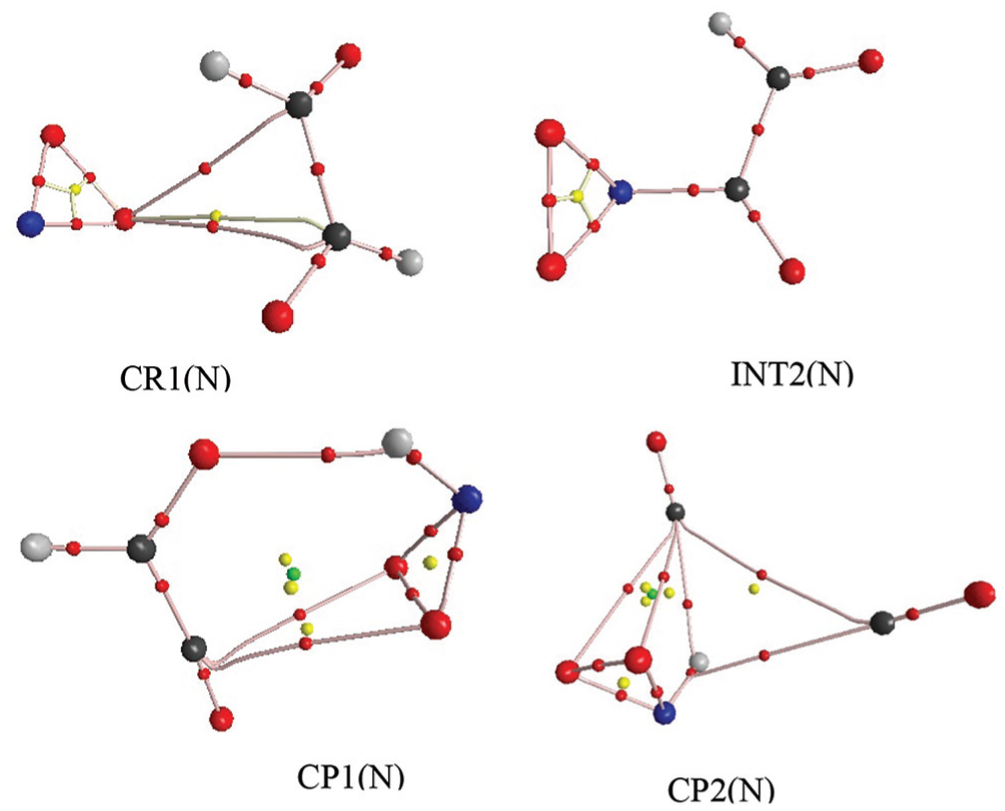
BCPs and bond paths for certain bonds of CR1(N), INT2(N), CP1(N) and CP2(N) in glyoxal + NO2 radical reaction at M06-2X level.
The CR2(N) species is produced by performing again reaction between NO2 and HC2O2 at the M06-2X level. The characterization of the covalent bond that is formed between C1 and N1 has also been reported in Table 1. Via the hydrogen transfer between the HC2O2 and NO2 radicals, a conversion of CR2(N) to CP2(N) takes place. Then, the CP2(N) species, with the weak van der Waals bonds, is easily converted to the final products. It is worth noting that while the CP1(N) structure at the M06-2X level is observed with two van der Waals and one hydrogen bond interactions, the same CP1(N) structure at the B3LYP level has one covalent, one van der Waals and one hydrogen bond interactions. Also, two and five van der Waals interactions are observed in the CP2(N) structure at the B3LPY and M06-2X levels, respectively.
However, as a complementary path, the optimization calculations for the species involved in the bent NO2 + glyoxal were also performed at the M06-2X/6-311++G(d,p) level (Figure 4).
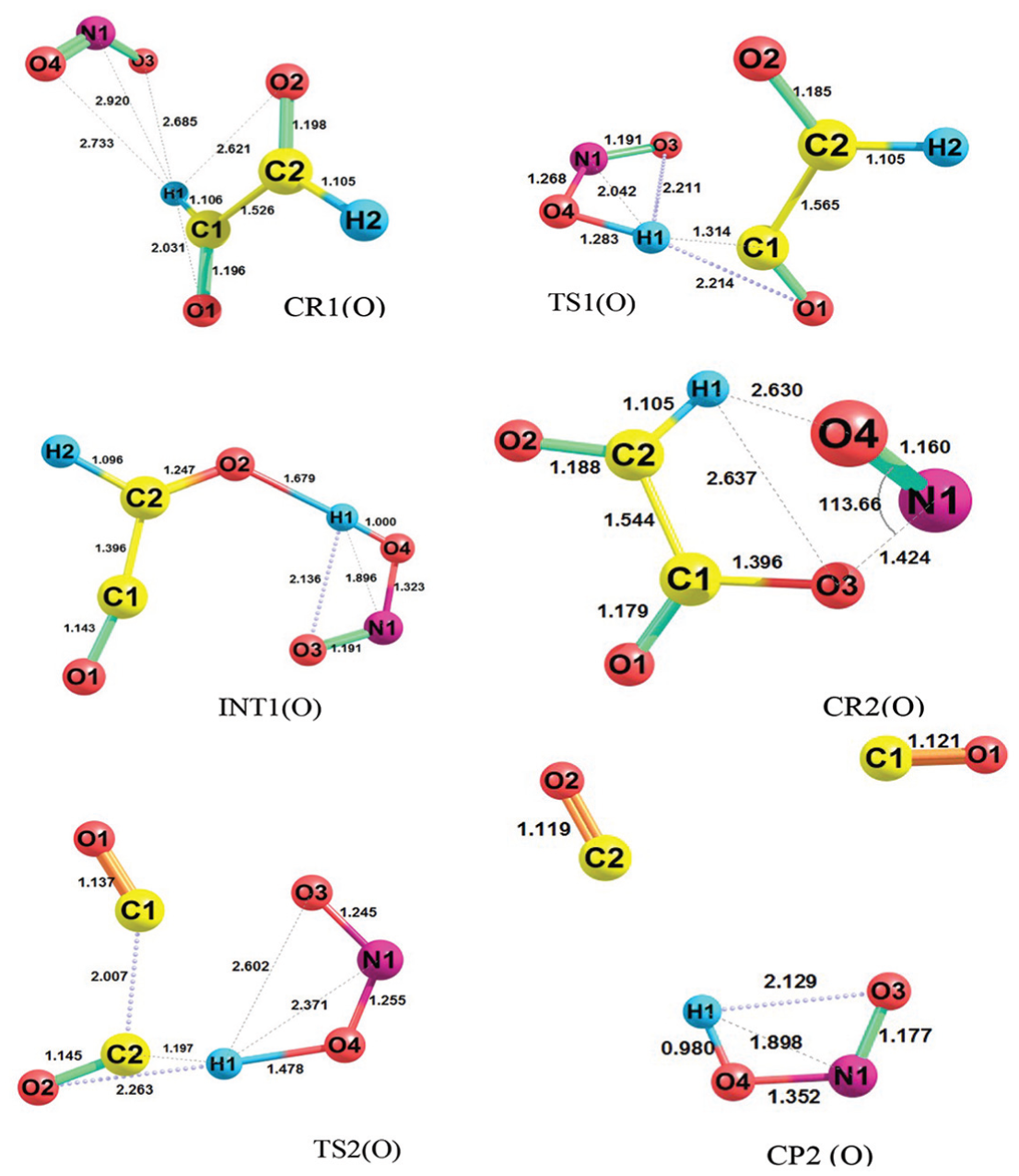
Geometries of all species involved in reactions of glyoxal + bent form of NO2 radical containing CR1(O), TS1(O), CP1(O), CR2(O), TS2(O) and INT2(O) at the M06-2X level (bond distances are in angstrom).
NBO analysis of glyoxal + NO2 reaction
NBO analysis is an appropriate method for studying interaction between two atoms (intramolecular) or molecules (intermolecular). This method provides a convenient basis for investigating charge transfer or conjugative interaction in molecular systems. Therefore, this method was used for studying charge transfer between glyoxal and NH2/NO2 radicals and their interactions. The NBO analysis has been performed on the CR, TS and CP molecules individually to identify and explain the interactions between glyoxal and NH2/NO2 radicals and charge transfers that occur inside and between species and to verify the accuracy of the results of AIM. The interaction between doubly occupied orbitals and unoccupied ones and the lowering of occupancy of Lewis-type orbital and delocalization energy correction is a very suitable guide to understand the molecular structure from the electronic point of view. The electron delocalization can be described as a charge transfer from a Lewis-type valence orbital (donor), with a decreasing of its occupancy, to a non-Lewis-type orbital (acceptor). 21 The larger E(2) values indicate the greater interaction between electron donors and acceptors. In these calculations, the intermolecular threshold of E(2) is 0.05 kcal mol−1.
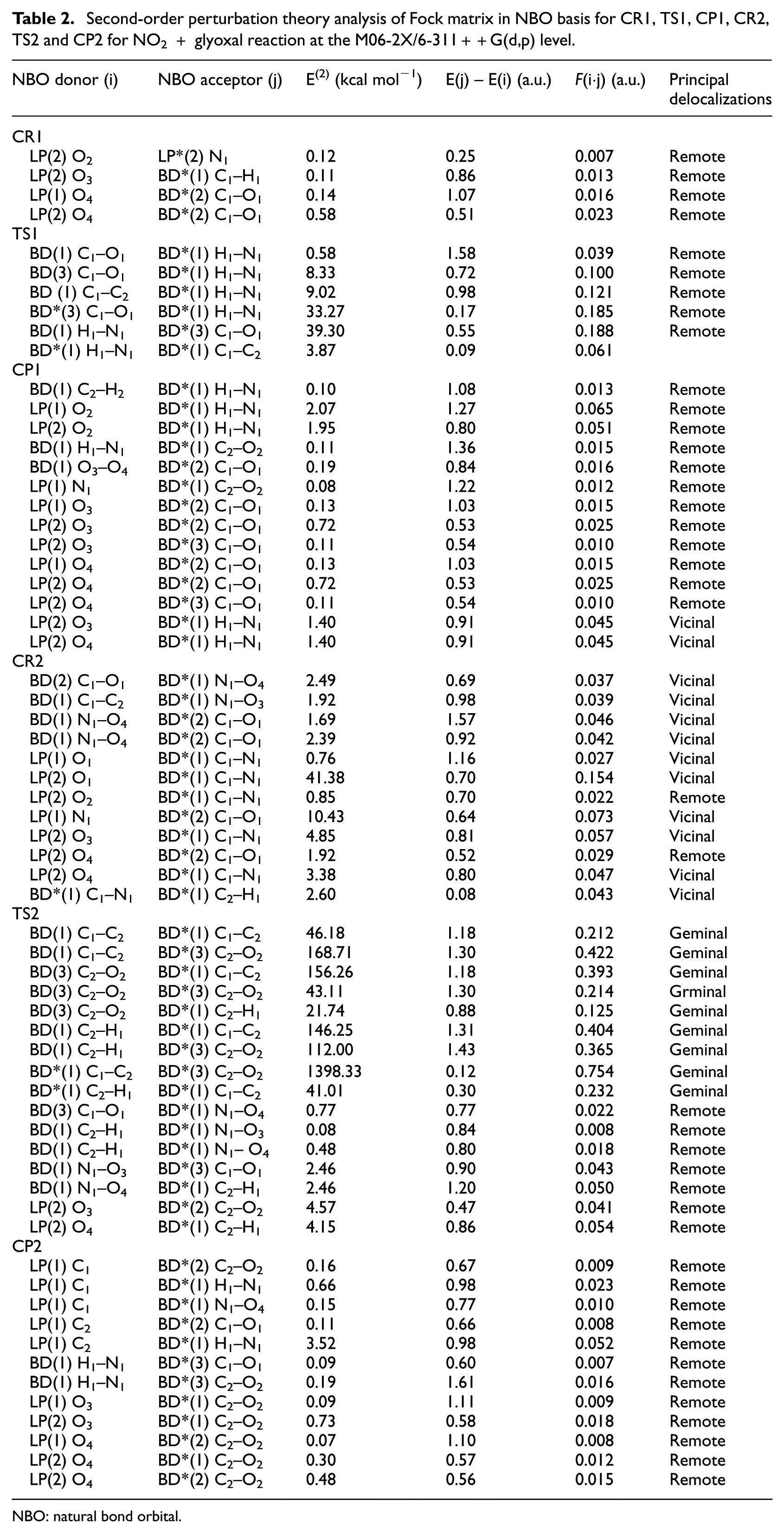
The E(2) energies and types of interactions of species involved in the glyoxal + NO2 reaction at M06-2X level in steps 1 and 2 are listed in Table 2. According to the results obtained, the E(2) values of CR1 indicate the greatest lowering of energy and the most stability occurs with the transfer of an electron from the lone pairs of one oxygen atom of NO2(O4) to the anti-bonding orbital of C1–O1 of glyoxal as an acceptor. These data also confirm the results of the AIM calculations. That is, as a result of NBO calculations of CR1, the O4 lone pair (NBO 27 in output of NBO calculation of CR1) is seen to be the lowest occupancy (0.914 electrons) and the highest energy (–0.477 a.u.) Lewis-type NBO and to be primarily delocalized into the C1–O1 anti-bond (a remote delocalization). Also, the results show that the Lewis-type NBOs in the NO2 unit of the CR1 describe about 98.1% of the total electron density, with the remaining non-Lewis-type density found primarily in the valence-shell anti-bonds (particularly, the anti-bonding orbital of C1–O1). The NBO results for TS1 with several remote delocalizations that have been brought in Table 2 confirm the estimated TS1 structure as well as hydrogen transfer to the N atom of NO2. The NBO investigation of this structure shows the existence of two individual units, C2HO2 and NHO2. The Lewis-type NBOs describe about 97.4% and 99.1% of the total electron density in C2HO2 and NHO2, respectively. These data can confirm the interactions between these two units and confirm the structure of the TS1. The investigation of NBO analysis of CP1 confirms the interactions generated by AIM calculations and shows the most delocalization stability can be achieved by electron transfer from lone pairs of the O2 atom of the C2HO2 unit to anti-bonding orbitals of the N1–H1 bond producing a hydrogen bond. All the remote delocalizations relating to CP1 in Table 2 confirm the six-membered ring structure obtained from the AIM calculation, but there are two extra vicinal delocalizations in Table 2 (relating to CP1) that show the presence of a covalent bond N–H in the NHO2 unit of CP1. The E(2) energies and types of interactions of step 2 of the NO2 + glyoxal reaction are also presented in Table 2. From this table, most data concerning CR2 indicate vicinal delocalizations and it can be clearly understood from them that there is a covalent bond C1–N1, indicating NO2 is bonded to carbon in the second step of the reaction and then the second hydrogen is transferred. The NBO data for TS2 are featured in Table 2. These amounts confirm the correctness of the estimated structure for TS2 because those confirm the van der Waals interactions in TS2. In addition, the remote delocalizations testify to the transfer of the second hydrogen atom to the NO2 unit of the TS2 structure. All the remote delocalizations obtained from the NBO results of CP2 (Table 2) confirm the van der Waals interactions obtained from the AIM calculations. From the small values of E(2), it can be concluded that the units in the complex can be easily separated to 2CO and NHO2.
Second-order perturbation theory analysis of Fock matrix in NBO basis for CR1, TS1, CP1, CR2, TS2 and CP2 for NO2 + glyoxal reaction at the M06-2X/6-311++G(d,p) level.
NBO: natural bond orbital.
Comparison of the results of NBO calculations at M06-2X and B3LYP levels shows that the B3LYP level, probably because of the basis set truncation errors, cannot reveal many interactions in the results and also, the E(2) values, in similar cases, at the B3LYP level are much higher than the same results at M06-2X level (nearly twice).
Gyloxal + NH2 reaction
In this section, all optimized geometries involved in NH2 + glyoxal reaction at the M06-2X level of DFT are shown in Figure 5. The creation of the final products of the reaction of NH2 and glyoxal, CO and NH3, can be represented in two steps. So, the mechanism of the first and the second hydrogen transfers can follow the equations below
The geometries of all species involved in the two steps of reaction containing CR1, TS1, CP1, CR2, TS2 and CP2, optimized at M06-2X/6-311++G(d,p) level, are presented in Figure 5. The CR1 is created when the NH2 approaches to the glyoxal with its N atom, and the distance between the nitrogen atom and the hydrogen atom of glyoxal is 2.422 Å at the M06-2X level of theory. The length of the N1–H1 bond calculated at the M06-2X level is 0.070 Å shorter than that given by the B3LYP level. Similar to the mechanism described for NO2 + glyoxal reaction, the complex of reactants of this reaction is converted into the complex of products by passing through its TS. In the CP1 structure, one bond containing O2–H3 with the bond length equal to 2.209 at the M06-2X level is observed. However, the results show that the bond length of N1–H1 in the CR2 structure at the M06-2X level is 2.575 Å. The point to note about CR2 is that the complex has a covalent bond in N1–C1 with the length of 1.360 Å. It is actually a covalent amide and a thermodynamic sink. Thus, CR2 to CP2 thermal conversion is highly unlikely, as the energy profile in Figure 7(b) shows (with the energy barrier of about 60 kcal mol−1). This conversion (CR2 → CP2), due to its high energy barrier, is completely thermally inaccessible unless it occurs in vibrationally activated CR2 before collisional deactivation. To our knowledge, a photochemical process can provide the required energy to achieve the vibrationally activated state of CR2. So, the photochemical transformation of CR2 to CP2 is possible and the reaction can progress in the activated vibrational modes. Also, CP2 formation from CR2 can, in turn, lead to final adducts, being more favourable from the thermodynamic point of view (ΔG° = –97.95 kcal mol−1). With precise investigation of the CP2 structure at the M06-2X level, it can be understood that the bond lengths of N1–C1, H1–C2 and C1–C2 are 3.048, 2.701 and 3.495 Å, respectively. These values of bond lengths reflect the interactions that have mostly van der Waals nature and that are confirmed by the AIM analysis. The values of the AIM topological parameters for certain bonds of species involved in the reaction pathway of CP1 and CP2 formation at the M06-2X level of DFT are illustrated in Figure 6. In this reaction, like the NO2 and glyoxal reaction, the bond lengths calculated at the M06-2X level are shorter than the corresponding bonds at the B3LYP level.
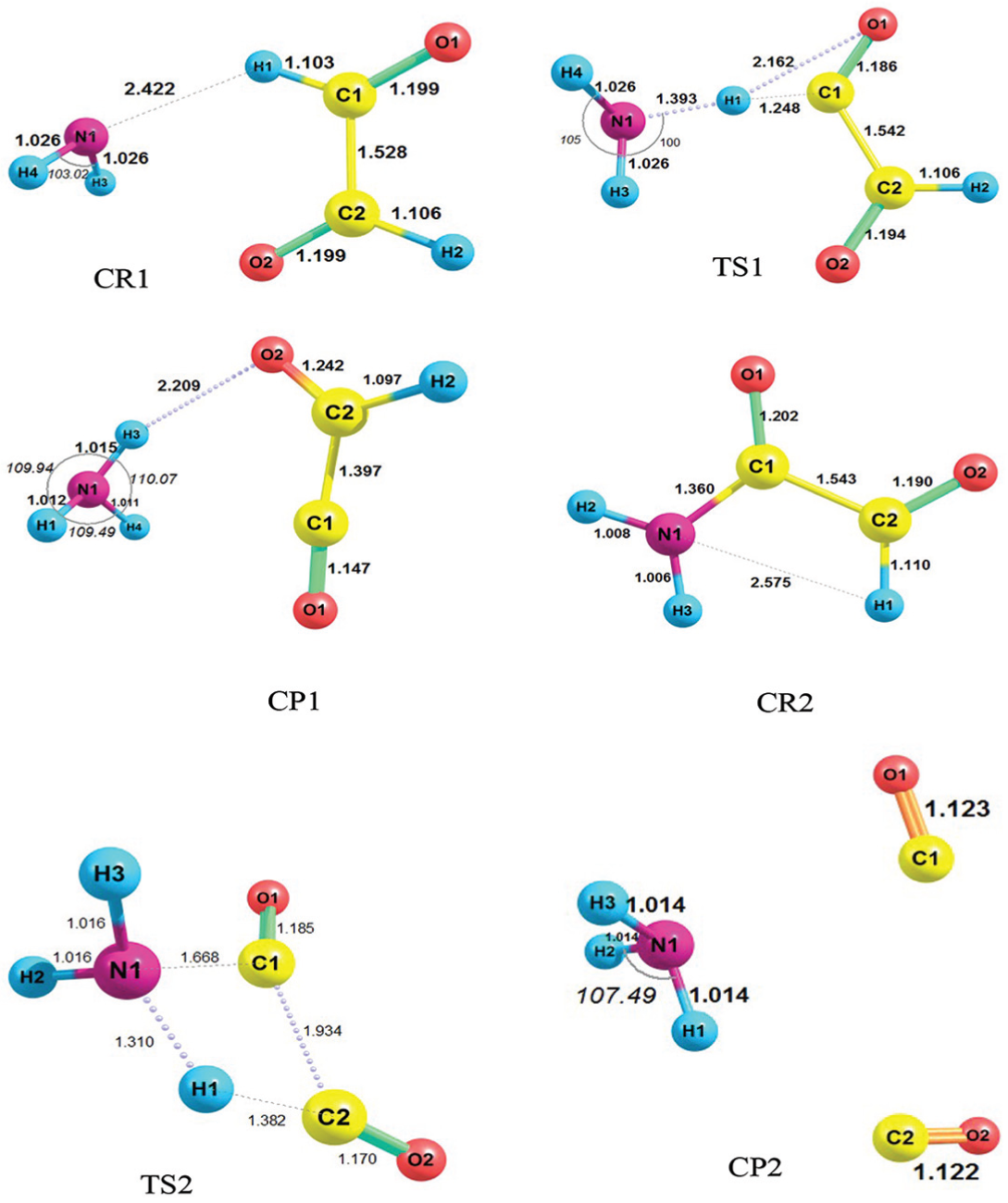
Geometries of all species involved in the reaction of glyoxal + NH2 radical containing CR1, TS1, CP1, CR2, TS2 and CP2 at the M06-2X level (bond distances are in angstrom).
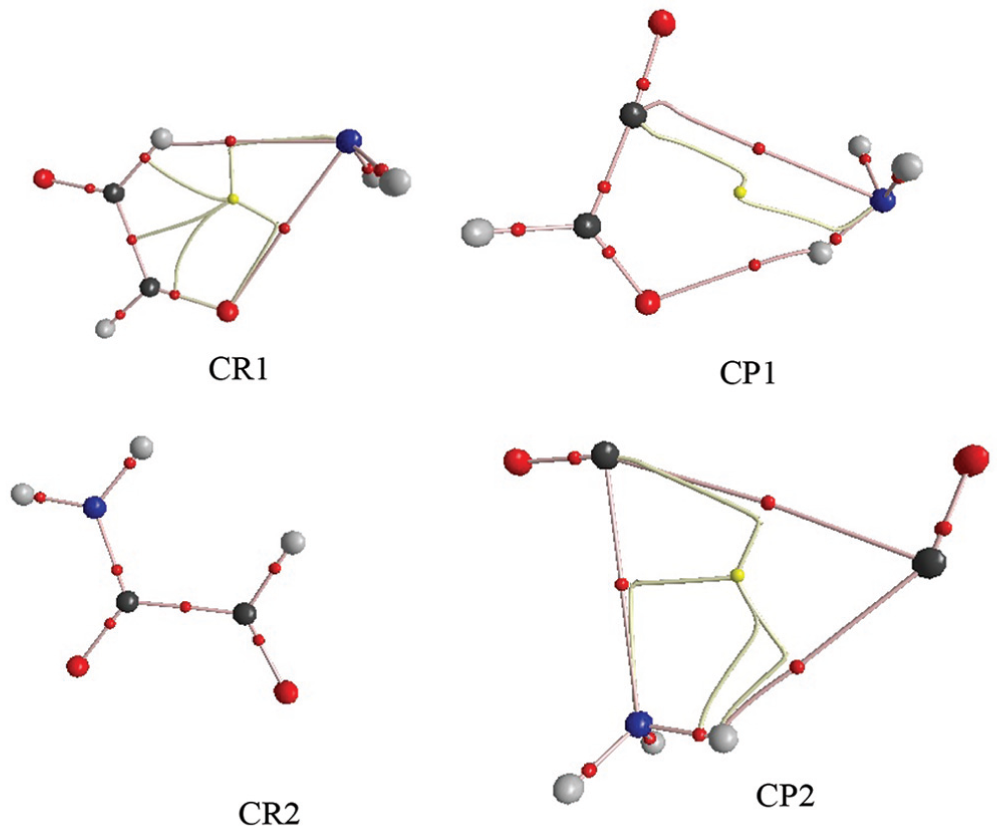
BCPs and bond paths for certain bonds of CR1, CR2, CP1 and CP2 in the glyoxal + NH2 radical reaction at the M06-2X level.
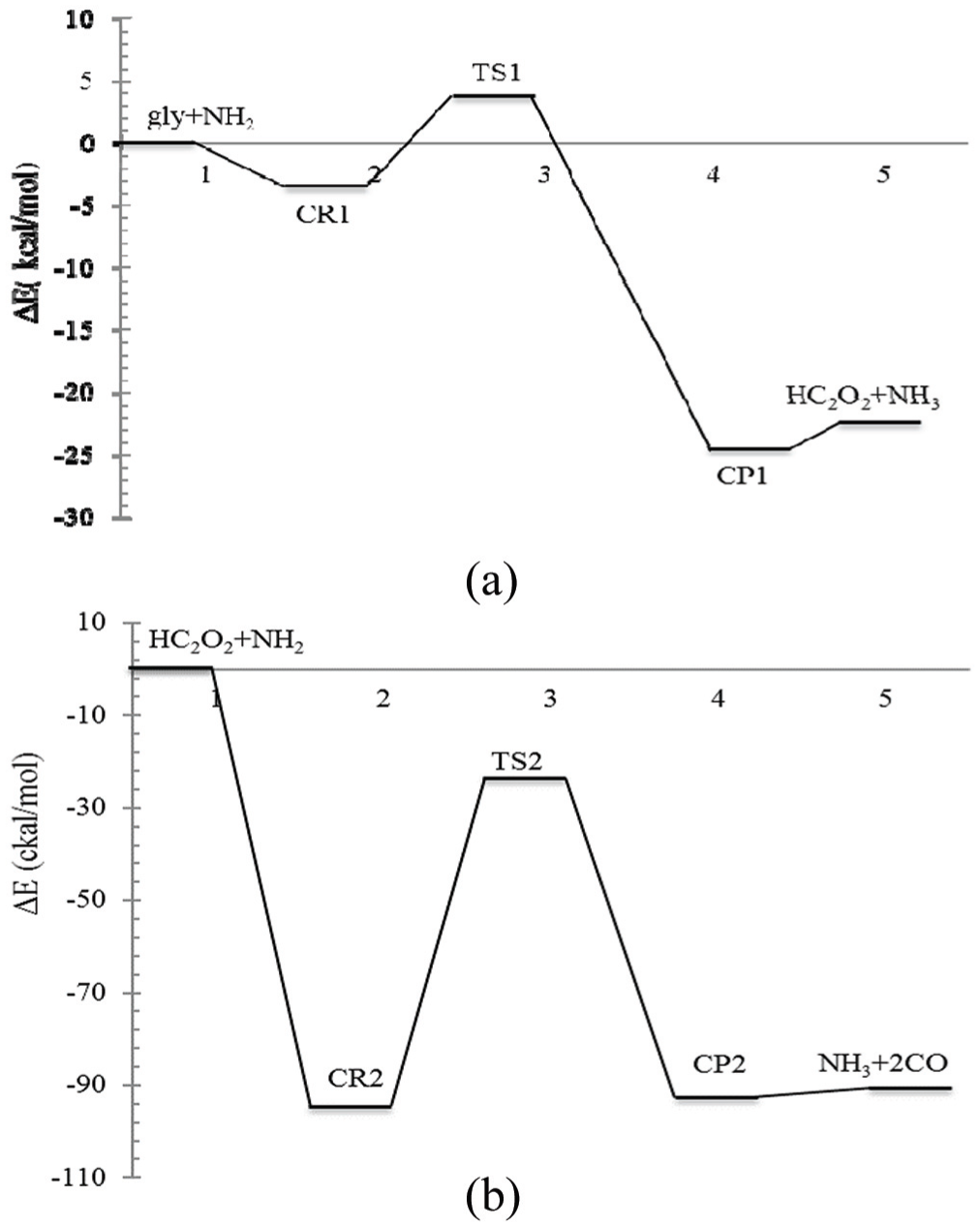
Potential energy profiles of the glyoxal + NH2 reaction: (a) first hydrogen abstraction and (b) second hydrogen abstraction.
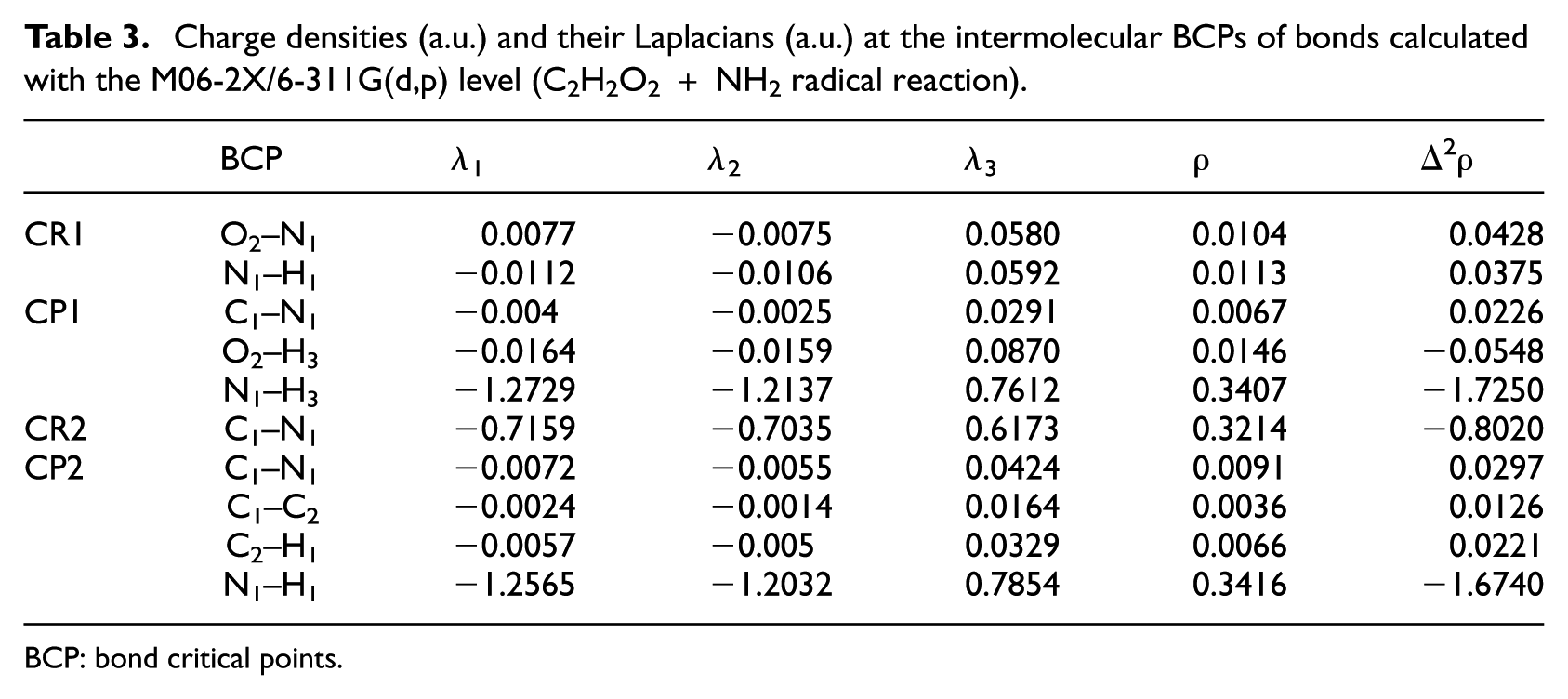
As mentioned previously, the AIM theory, due to the correlation between the bond strength and the charge density (ρ) in the BCP, is appropriate to explain the nature and strength of bonds. According to the AIM analysis results (shown in Table 3), it has been proven that there exist two BCPs in the CR1, one is situated between O2–N1 (
Charge densities (a.u.) and their Laplacians (a.u.) at the intermolecular BCPs of bonds calculated with the M06-2X/6-311G(d,p) level (C2H2O2 + NH2 radical reaction).
BCP: bond critical points.
In this reaction, what is noted as the CP2 structure will decompose to 2CO and NH3 when the hydrogen atom transfers from the carbon atom to the nitrogen atom and van der Waals bonds break. However, it is worth mentioning that the hydrogen transfer is done completely by formation of a N1–H3 bond in the CP1 structure. The AIM analysis shows that this bond is covalent in nature. The CR2 is also converted to CP2 via an intramolecular hydrogen transfer. By looking at Table 3, we found that as a result of this transfer, another covalent bond is created (N1–H1). An important difference between two levels which were studied is the different interactions in the CR1 structure. At B3LYP, one hydrogen atom of NH2 connects with an oxygen atom of glyoxal and creates a six-membered ring structure CR1, while at the higher level of theory, CR1 has a five-membered ring structure and H3 is not in the ring (see Figure 6).
NBO analysis of glyoxal + NH2
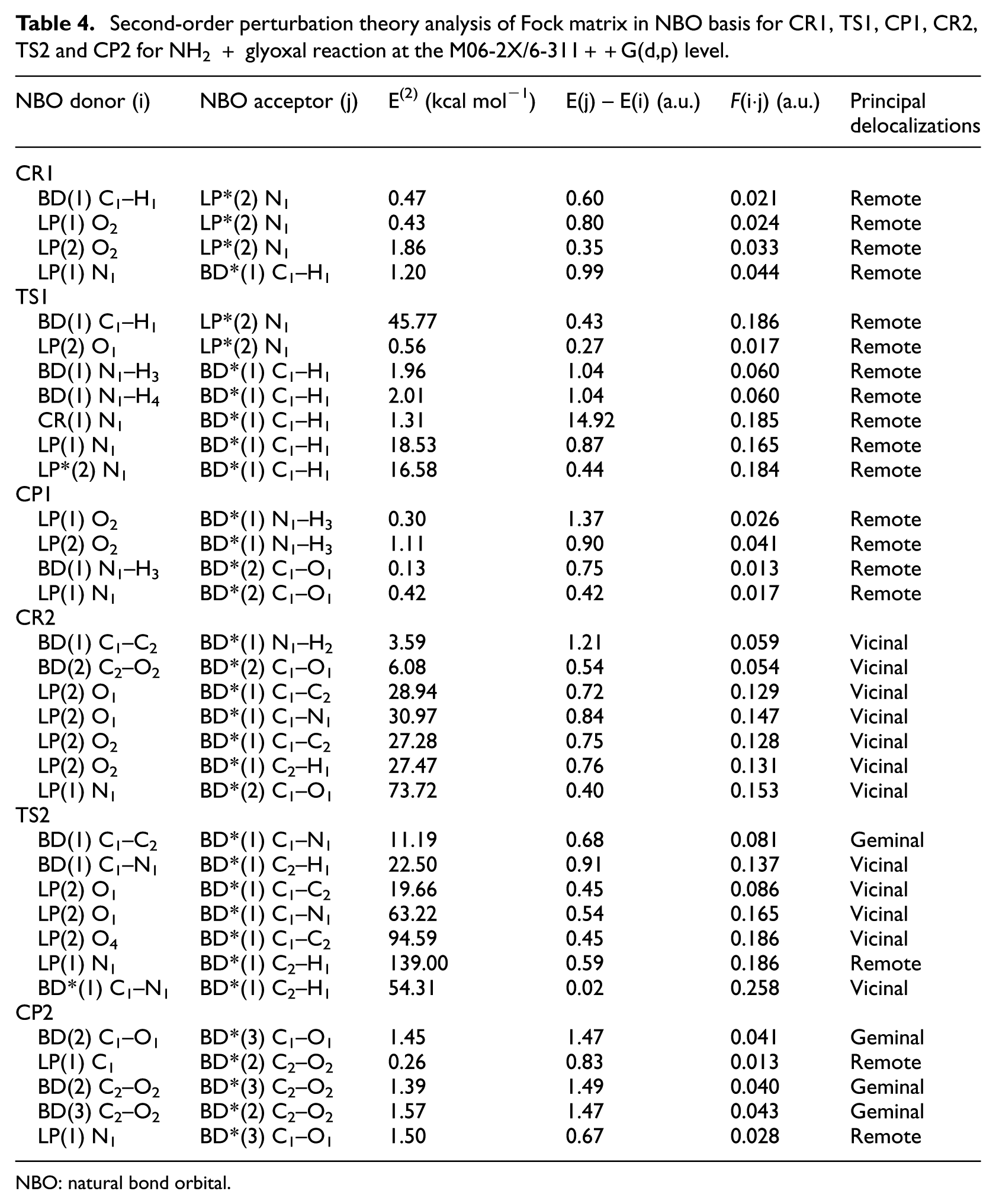
The important details of the NBO analysis for structures related to the NH2 + glyoxal reaction at the M06-2X level, CP1, TS1, CR1, CP2, TS2 and CR2 are presented in Table 4. These results confirm what is obtained in the AIM calculations at the M06-2X level, that is, the five-membered ring structure for CR1. The strongest donor–acceptor interactions are between N1 of NH2 and O2 of glyoxal, and neither of two hydrogen atoms of NH2 has a significant interaction. As shown in Table 4, the greatest delocalization energy in TS1 is related to the overlap of non-bonding electron LP(1) N1 and anti-bonding BD*(1) C1–H1 with E(2) equal to 18.53 kcal mol−1. The results in the table show well that the hydrogen atom of glyoxal transfers to the NH2 radical. However, the NBO analysis of the CP1 at the M06-2X level obviously displays the evidence of the electrostatic interaction between O2 lone pairs and the anti-bonding orbital BD*(1) N1–H3, having a delocalization energy of 0.30 and 1.11 kcal mol−1 for two lone pairs. These remote delocalizations indicate CP1 easily breaks down to the products of the first stage of reaction. During the second step of reaction, at the M06-2X level, the most stabilization energy E(2) of CR2 is related to a hyperconjugative interaction BD(1) C1–C2 and BD*(1) N1–H2 amounting to 1.21 kcal mol−1. The vicinal delocalizations in CR2 represent the covalent nature of the bonds of this complex. CR2 converts to CP2 via TS2. It should be noted that an important contribution of electron transfer to TS2 is given by interaction of LP(1) N1 and BD*(1) C2–H1. The value of this contribution is 139.00 kcal mol−1. The nature and strength of the inter- and intramolecular bonding can be explored by studying the E(2) values of the interactions. Here, the nitrogen lone pair is seen to have the lowest-occupancy (1.742 electrons) and highest-energy (–0.407 a.u.) Lewis-type NBO and to be primarily delocalized into the anti-bonding orbital C2–H1. According to the results, the energy values of electron transfer (E(2)) of inter- and intramolecular interaction between the units of the all species that were calculated at the M06-2X level are larger than those calculated at the B3LYP level. In CP2, the small values of delocalization energies (E(2)) state that the interactions are van der Waals, and this structure breaks to final products, NH3 and CO.
Second-order perturbation theory analysis of Fock matrix in NBO basis for CR1, TS1, CP1, CR2, TS2 and CP2 for NH2 + glyoxal reaction at the M06-2X/6-311++G(d,p) level.
NBO: natural bond orbital.
Profiles of energies and thermodynamic results of NH2 + glyoxal and NO2 + glyoxal reactions
To get a deeper insight into the NH2/NO2 + glyoxal reactions, their profiles were investigated at B3LYP and M06-2X levels of DFT. The energies of all species involved in the reactions were obtained relative to the separated reactants by computing the total electronic energies and zero-point vibrational energy corrections (ZPE) for all the stationary points at 298.15 K. The results of these energies for step 1 of the NH2/NO2 + glyoxal reaction at the M06-2X level are plotted in Figures 7(a) and 8(a), respectively. However, the total (Figures 7 and 8) energies and corresponding relative energies for the reactants of NH2 + glyoxal and NO2 + glyoxal at the M06-2X level are presented in Tables 5 and 6, respectively. For the first step of both reactions, the overall energy barrier values (ETS−ΣEreactants) are expected to be about 20.90 and 3.67 kcal mol−1 for the glyoxal + NO2 and glyoxal + NH2 reactions, respectively. Similar to the first steps of both reactions, the energy profiles for the second H abstraction from the residual fragment of glyoxal (HC2O2) with NH2/NO2 are plotted in Figures 7(b) and 8(b), respectively. The results show that the total energy barriers of the second step of NO2 and NH2 reactions are 16.71 and −23.70 kcal mol−1 (i.e. barrier less), respectively. As seen in Figures 7 and 8, the energy barrier of the first step of the glyoxal + NH2/NO2 reaction is lower than that of the second. So, the first step of these reactions is faster than the second and it can be considered that it is favourable from the kinetic viewpoint. However, in both reactions, the CR2s are located at the bottom of deeper wells than are the CR1s, maybe because the CR2s are created by interaction of two radical species.
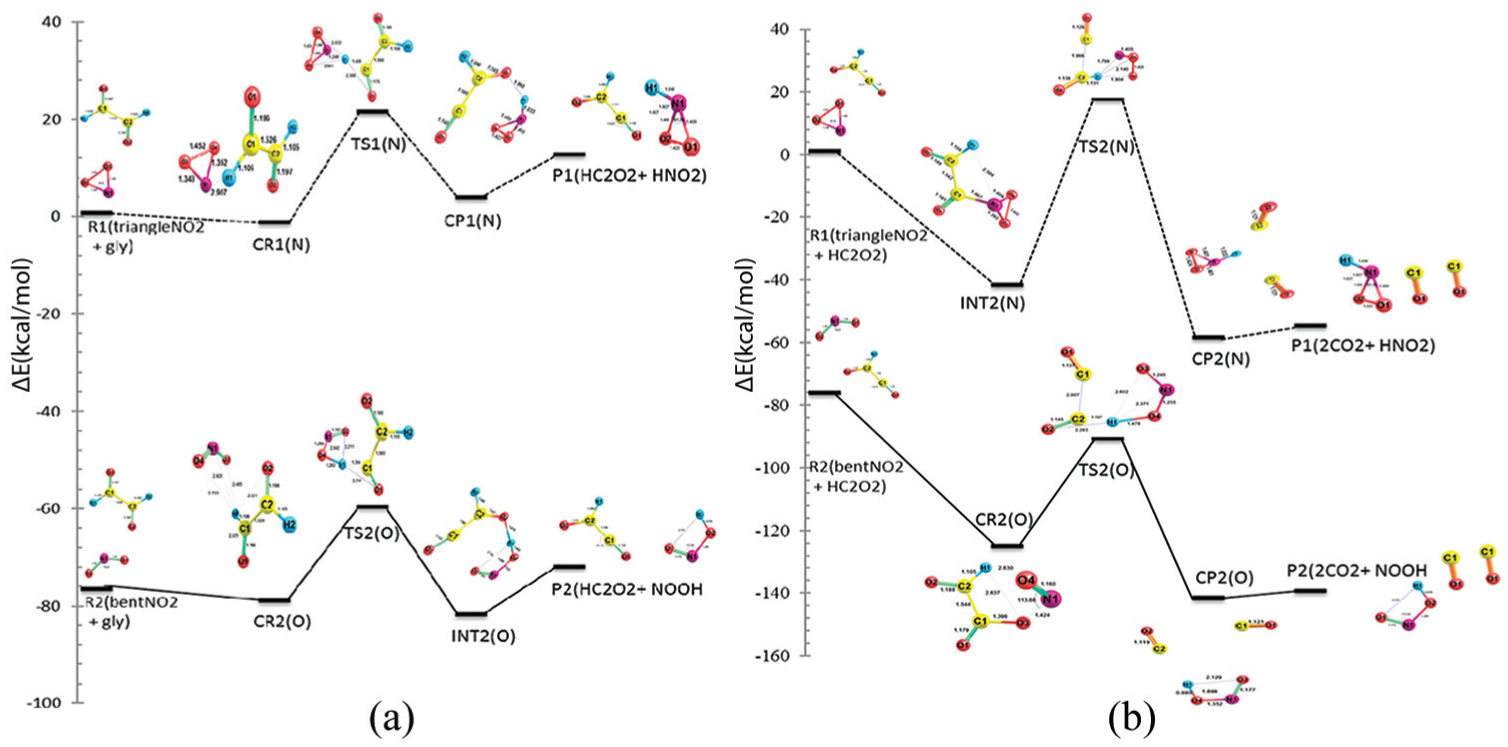
Potential energy profiles of the paths of glyoxal + NO2 reaction: (a) first hydrogen abstraction and (b) second hydrogen abstraction.
Total energies (Eelec + ZPE in Hartree) and relative energies (kcal mol−1) of species involved in the glyoxal + NH2 and HC2O2 + NH2 reactions at the M06-2X/6-311++G(d,p) level.
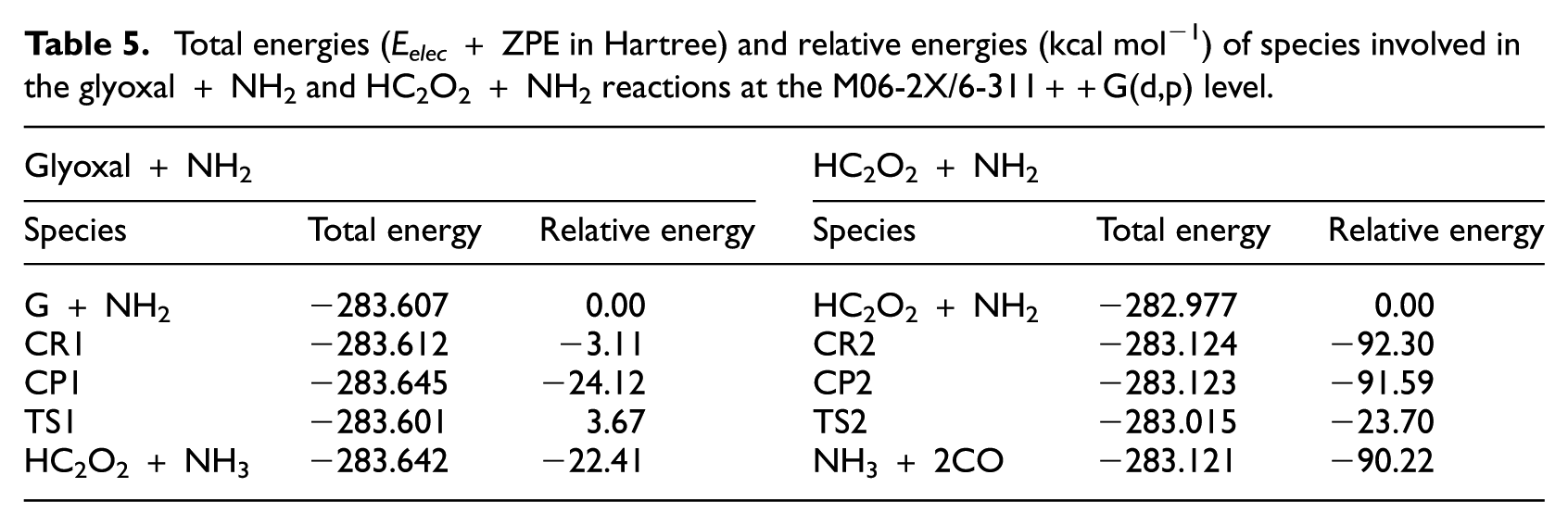
Total energies (Eelec + ZPE in Hartree) and relative energies (kcal mol−1) of species involved in the glyoxal + NO2 and HC2O2 + NO2 reactions at the M06-2X/6–311++G(d,p) level.

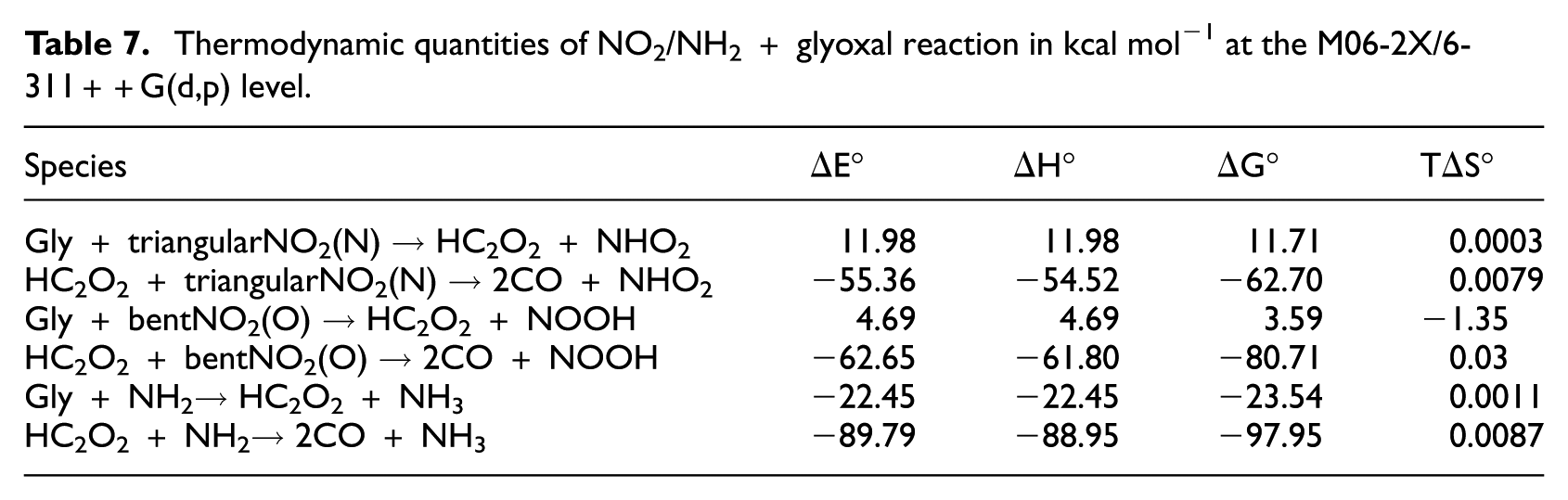
The thermodynamic quantities of all the reactions at the M06-2X level are presented in Table 7. It can be concluded that the first steps of the reactions, glyoxal + NO2 and glyoxal + NH2, are estimated as endothermic and exothermic, respectively, while the enthalpy values at both levels show that the second steps of the two reactions are exothermic. These results also include the free energy values. The free energy values of glyoxal + NO2 and glyoxal + NH2 with regard to the first steps of the reactions at the M06-2X level are 11.71 and −23.54 kcal mol−1, respectively. This means that the first step of glyoxal + NH2 reaction is thermodynamically satisfactory. However, both the HC2O2 + NO2 and HC2O2 + NH2 reactions have negative values of their free energy. The magnitude of free energy of HC2O2 + NH2 is about 35.25 kcal mol−1 more negative than that of HC2O2 + NO2. It is noteworthy that from the thermodynamics point of view, the whole reaction of glyoxal + NH2 is more favourable than the whole reaction of glyoxal + NO2. The thermodynamic results of the two reactions at the B3LPY level are different from those obtained at the M06-2X level. The values of (ΔHM06-2X−ΔHB3LPY) for glyoxal + NO2 and glyoxal + NH2 for the first steps of the reactions are −0.23 and 1.07 kcal mol−1, respectively. Thus, the enthalpy change of the first reaction step of glyoxal + NO2 at the M06-2X level is lower than that at the B3LPY level, while for glyoxal + NH2, the enthalpy change at the M06-2X level is larger than that at the B3LPY level. The results show the second steps of both reactions at the M06-2X level are more exothermic (about −4.53 kcal mol−1 for H2C2O2 + NO2 and −10.23 kcal mol−1 for HC2O2 + NH2) than that at the B3LPY level. By comparing the free energy values obtained from B3LPY and M06-2X levels, it can be commented that the progress of the both reactions at the M06-2X level is more favourable thermodynamically than that at the B3LYP level.
Thermodynamic quantities of NO2/NH2 + glyoxal reaction in kcal mol−1 at the M06-2X/6-311++G(d,p) level.
Rate constants
In kinetic study, the rate constant is a key parameter. It can be computed from the Eyring equation using transition state theory (TST) based on statistical mechanics formalism. 22 To apply TST to bimolecular reactions, the accurate transition structure and also the partition functions of the internal degree of freedom (electronic, transitional, vibrational and rotational partition functions) are needed.
The Eyring equation is familiar for calculating the rate constants of bimolecular reactions; it can be written as follows

where
In this study, we used the Eckart correction 23 for the reaction rate, which is related to the value of the imaginary frequency, νim, on the minimum energy path of the reaction, from reactants to TS to products. The tunnelling factor in this work is computed as follows

where νim is used for the imaginary value of the frequency and E0 is the potential energy at the TS.
The rate constants of the reactions have been calculated at the M06-2X/6-311++G(d,p) level for all the energy pathways of glyoxal + NO2/NH2 reactions, and these are shown in Figures 9 and 10.
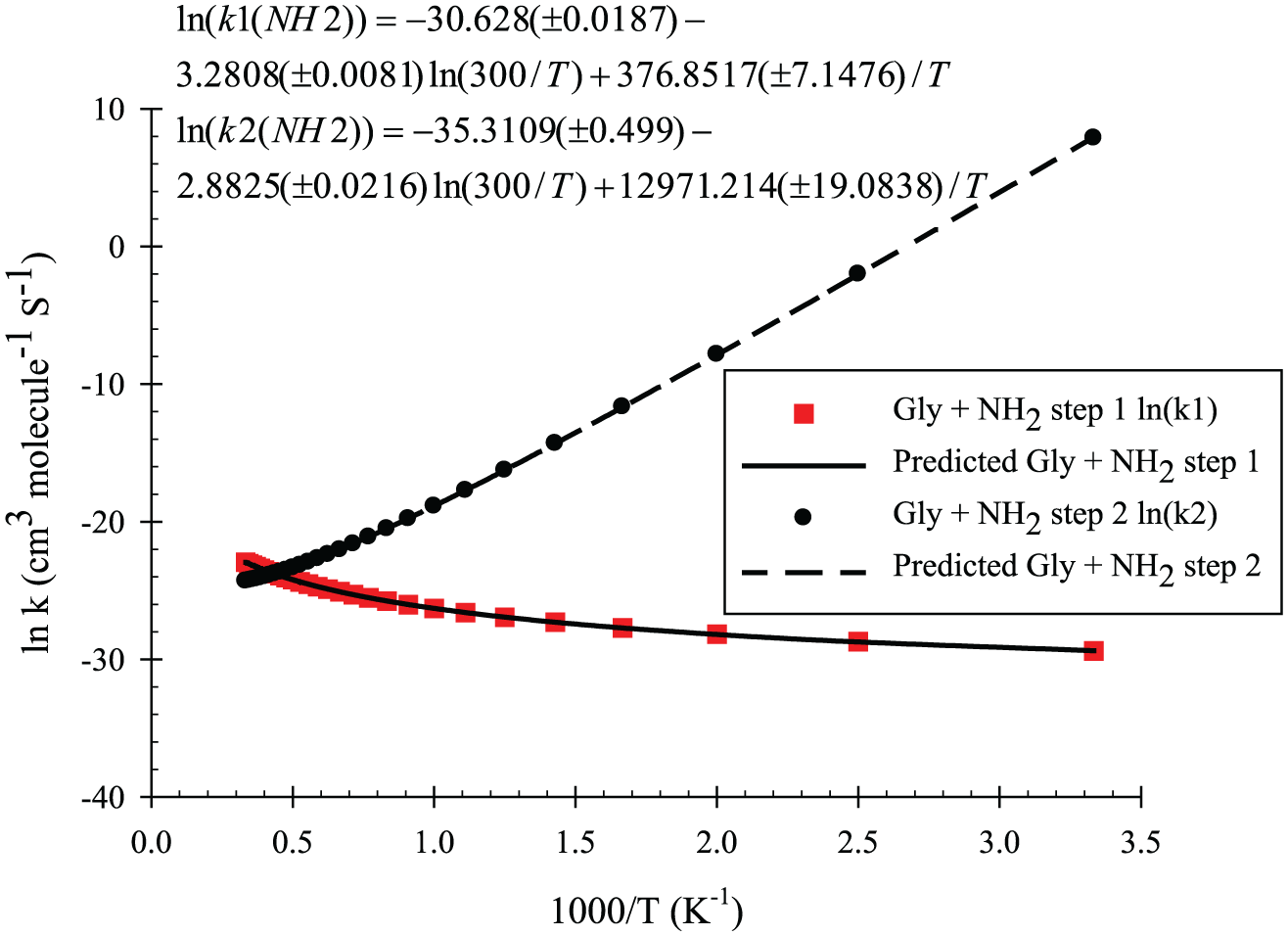
ln k vs (1000/T) plotted for NH2 + glyoxal at the M06-2X/6-311G(d,p) level.
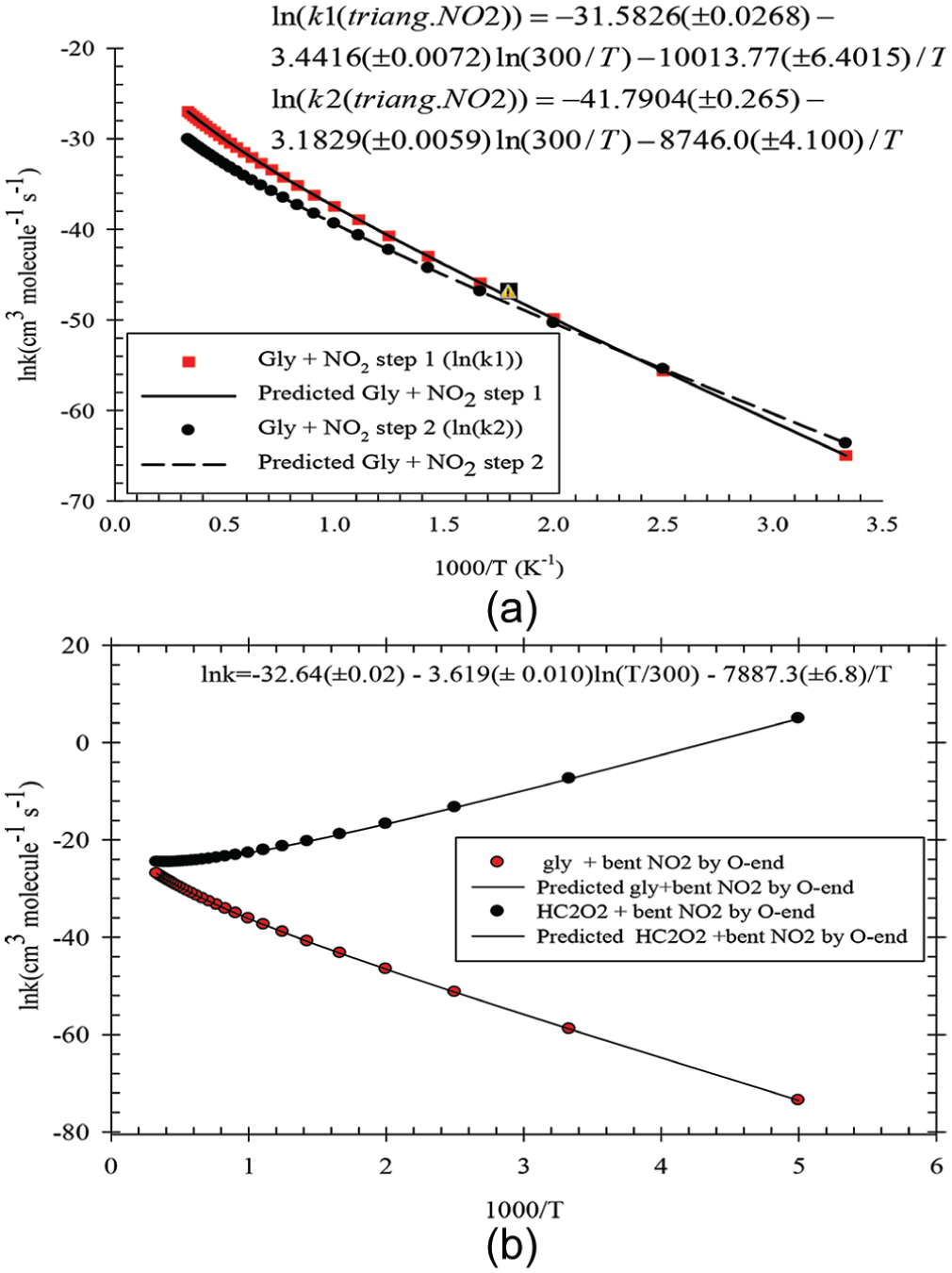
ln k vs (1000/T) plotted for NO2 + glyoxal at the M06-2X/6-311G(d,p) level: (a) triangular conformer and (b) bent conformer of NO2.
Comparison of our results at the two calculated levels for the NH2 + glyoxal and NO2 + glyoxal reactions shows that the M06-2X results are better because the predictions of M06-2X about the rate constants involve low computational cost, especially for large or moderate size systems; accordingly, we selected them for our discussions. Based on the accuracy of the calculated levels for title reactions, the M06-2X results are plotted in Figures 9 and 10 and the B3LYP results are collected in Supplemental Table 4S of supporting information. Figure 9 shows our calculated results for the first and the second hydrogen abstractions for glyoxal + NH2, and Figure 10 shows the same results for the first and the second hydrogen abstractions for glyoxal + NO2 (Figure 10(a) for glyoxal + triangular conformer of NO2, and Figure 10(b) for O-end of NO2). All the results of the mentioned reactions show non-Arrhenius behaviour, thus they are related to the role of the hydrogen shift in the reaction mechanisms. Also, in the NH2 + glyoxal reaction, because there is a TS with lower energy than that of the reactants (this reaction is so-called the barrier-less reaction), the rate constant of this reaction decreases with increasing temperature.
From the calculated rate constants in the NH2 + glyoxal reaction, it can be understood that with increasing temperature, the rate constant of the second path decreases (barrier-less process), while that of the first path increases. So, by our prediction, the first and the second paths are important at high and low temperatures, respectively. At moderate temperatures, a competition exists between the two pathways.
For the NO2 + glyoxal reaction, the energy of TSs in the two steps of hydrogen abstraction is greater than those of the reactants. So, the rate constant increases with increasing temperature. As we can see in Figure 10(b), at low temperature, the second mechanism is more significant than that of the first. But this figure shows when the temperature increases to moderate or high values, the rate constant of the first H abstraction step is more significant than that of the second one and it is related to the energy barriers of their TSs (see Figure 8).
The rate constants were calculated for the mentioned pathways using TST for bimolecular steps as implemented in the GPOP programme. 24 The calculated rate constants are improved by the Eckart correction method for tunnelling. The values of the rate constants for all steps of both reactions (for ease of use) are fitted in the three-term expressions of the following type
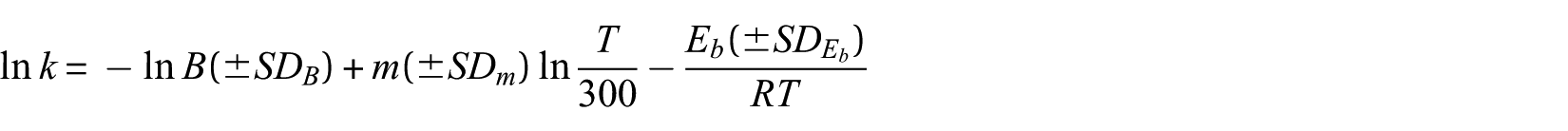
where B, m and Eb are the adjustable parameters, SDi (i = B, m and Eb) is the standard deviation and T is the absolute temperature. The fitted equations are reported as

The Arrhenius activation energy can be calculated as 22 from the equation Ea = Eb + mRT. The Arrhenius activation energies are 5.04 and −100.65 kJ mol−1 for the first and second steps of the glyoxal + NH2 reaction, respectively, and those of Gly with cyclic and bent NO2 reactions are 91.83 and 74.60 kJ mol−1 for the first step and 80.65 and 76.58 kJ mol−1 for the second step, respectively. The activation energy with respect to the most stable structure of NO2 (bent form) as an original reactant for the overall reaction of the cyclic structure can be calculated from the equation Ea overall = Ea + ΔH0, where ΔH0 is the standard enthalpy of NO2(bent) → NO2(cyclic) transformation. Due to the most stable form of the NO2 molecule, the overall activation energy for the first and second steps of reaction are 414.82 and 397.59 kJ mol−1, respectively. These values of the activation energies can be achieved by light at about 300 nm in wavelength that is available in the atmosphere.
Conclusion
In this study, the reaction pathways of glyoxal with NO2 and NH2 have been characterized with the B3LYP and M06-2X levels of theory in conjunction with the 6-311++G(d,p) basis set. The results show that in both reactions, the hydrogen transfer takes place over two steps. By starting from the CR1 species in both reactions with the same mechanism at the B3LPY and M06-2X levels, the different structures for reactant and product complexes and TSs have been considered. The conclusions can be summarized as follows:
The calculated results show that all newly formed bonds during the two steps of the NH2/NO2 + glyoxal reactions at the M06-2X level are shorter than those from the B3LPY level. This corresponds with the lower energy value for the species in the reaction coordinate. So, for the study of intermolecular interactions, the M06-2X level is better in comparison with the B3LYP level as we might expect.
The formation of the CR1 in the NO2 reaction depends on electron transfer from the lone pairs of an oxygen atom of NO2 to the anti-bonding C–O orbital of the glyoxal, while in the NH2 reaction, the formation of this complex is due to donation of the lone pair of the nitrogen atom of NH2 to the anti-bonding C–H of the glyoxal.
By comparing the E(2)s of NBO calculations at the M06-2X level for the two CR1s of both reactions, it can be concluded that stability and delocalization energy in the CR1 of NH2 are more than that of NO2 (about 11 times). Probably, the electropositivity of H has made nitrogen in the NH2 radical to be the better donor. Also, the stability of this complex in comparison with the reactants in the reaction of NH2 and glyoxal is about 1.65 times higher than that of the ratio in the reaction of NO2 and glyoxal.
Studies of the CR2s show that in the second step of the NH2/NO2 + glyoxal reactions, at first, both radicals are placed on HC2O2, and then hydrogen exchange is performed. In fact, in the second step, the presence of a carbonyl group in HC2O2 provides a suitable position for substitution of radicals. In this way, the complexes are located in the bottom of the wells of potential energy. The depth of this well is higher in the reaction with NH2.
The results of AIM and NBO calculations displayed some of the interactions that are presented at the M06-2X level as van der Waals, while at the B3LPY level, the interactions obtained are covalent, and some of them are not observed at all.
The NBO results confirm the formation of involved structures in the estimated mechanism at the stationary points.
According to the activation thermodynamic results, it can be concluded that the glyoxal + NO2/NH2 reaction has lower barrier energy than the second step featuring HC2O2 + NO2/NH2. Also, the results of the free energy values obtained from B3LPY and M06-2X levels showed that the progress of both reactions at the M06-2X level is more favourable thermodynamically than that at the B3LYP level.
Supplemental Material
PRK1900773_ESI_word97 – Supplemental material for Atmospheric reactions of glyoxal with NO2 and NH2 radicals: Hydrogen abstraction mechanism and natural bond orbital analysis
Supplemental material, PRK1900773_ESI_word97 for Atmospheric reactions of glyoxal with NO2 and NH2 radicals: Hydrogen abstraction mechanism and natural bond orbital analysis by Homeira Saghafi and Morteza Vahedpour in Progress in Reaction Kinetics and Mechanism
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.