
Abstract
Using Fe6(OH)18(H2O)6 as a ring cluster model for superparamagnetic iron oxide nanoparticles, noncovalent configurations and three mechanisms of covalent functionalization of superparamagnetic iron oxide nanoparticles with cyclophosphamide an anticancer drug were studied. Quantum molecular descriptors, solvation, and binding energies of noncovalent interactions were investigated the in gas and solution phases at the B3LYP and M06-2X density functional levels. In the vicinity of superparamagnetic iron oxide nanoparticles, the reactivity of the drug increases, showing cyclophosphamide can probably bind to superparamagnetic iron oxide nanoparticles through Cl (k1 mechanism), P=O (k2 mechanism), and NH in a six-membered ring (k3 mechanism) groups. The activation parameters of all pathways were calculated, indicating the high barriers related to the k1 and k2 mechanisms are higher the barrier related to the k3 mechanism. The k3 mechanism is also spontaneous and exothermic and is therefore the preferred mechanism for covalent functionalization.
Keywords
Introduction
Superparamagnetic iron oxide nanoparticles (SNPs) consist of elements such as iron, cobalt, nickel, and their oxides which are increasingly utilized in different areas.1–4 Due to their unique chemical and electronic properties, these materials are employed in biological and pharmaceutical research.5–9 SNPs have a large surface-to-volume ratio, and for this reason, they could be used effectively to transfer different materials including therapeutic agents.10–16
The use of magnetism in medicine was introduced by Freeman et al. in the 1970s. 17 Since that time, much research has been done in this field, which has led to the design of different magnetic particles. Generally, drugs do produce some side effects, which normally arise from lack of precision and specificity in drug action. Targeted drug delivery for diseases such as cancer causes lower dosage of drugs to be required, consequently leading to reduction of side effects such as vomiting, breathing trouble, cardiotoxicity, and hair loss.18,19
The magnetic properties of SNPs make them suitable to be guided towards the intended site such as a cancerous tumor by an external magnetic field.5,20,21 The drug will be released in cancer tissues by factors such as enzymatic activity, changes in pH, temperature, and osmolality.22,23
Iron oxide can exist as different chemical compositions, such as magnetite (Fe3O4) and maghemite (γ-Fe2O3). These particles demonstrate the phenomenon of superparamagnetism. 24 SNPs are small synthetic Fe3O4 (magnetite), γ-Fe2O3 (maghemite), or α-Fe2O3 (hematite) particles.25,26 In the presence of an external magnetic field, SNPs become magnetized up to their saturation magnetization and, in the absence of an external magnetic field, they do not show any residual magnetic interaction. This property is dependent upon size, and generally increases when the diameter of the iron oxide nanoparticles is as low as 10–20 nm. Magnetite and maghemite have the widest usage in biomedical applications. Covalent and noncovalent (hydrogen bonds and van der Waals interactions) functionalizations25,27–30 play a principal role in drug delivery and biological applications using iron oxide nanoparticles. The possibility of targeted drug delivery causes reduction of the amount of drugs consumed and consequently the reduction of their side effects.31–33 This superparamagnetism, exclusive to nanoparticles, is very valuable for their application as a drug delivery vehicle because they can actually drag drug molecules to their target site in the body under the impact of an external magnetic field.34–36
Cyclophosphamide (CP) or cytophosphane, has anticancer activities and is effective in the treatment of lymphoma, leukemia, multiple myeloma, ovarian cancer, breast cancer, sarcoma, neuroblastoma, and small-cell lung cancer.37,38 The formulation of appropriate molecular models in solution (especially water) is of special importance in understanding the mechanistic behavior of drug delivery systems such as SNPs.
The most appropriate method for designing such models is quantum chemical calculations. In 2016, the chemistry Nobel Prize was awarded for the design and manufacture of molecular machines, an important application of which is in drug delivery systems.39–41
In this work, we have used quantum chemical calculations to analyze mechanisms of noncovalent and covalent functionalization42,43 of SNPs with cyclophosphamide drug needed. So far, little research has been done on the mechanism of functionalization of SNPs, and therefore, this work could inspire researchers in the design and manufacture of new drug delivery systems.
Computational method
All quantum chemical calculations have been done using the GAUSSIAN 09 package 44 at the B3LYP45–47 hybrid density functional level together with the 6-31G(d,p) basis set.48,49 For Fe atoms, the LANL2DZ basis set 50 with effective core potential functions was employed. We repeated the calculations for noncovalent functionalization using the M06-2X functional51,52 which implicitly accounts for “medium-range” electron correlation (systems separated by about 5 Å or less), 53 as M06-2X can describe the dispersion interactions within many systems. 54 Standard convergence criteria for geometry optimization were used. For all configurations, all degrees of freedom were optimized. The transition states obtained were confirmed to have only one imaginary frequency of the Hessian. The zero-point corrections were also used to calculate activation energies. To achieve higher accuracy, approximation methods such as ONIOM 55 were not used.
The solvent can have a significant effect on chemical systems and reactions explicitly56–61 or implicitly. The polarized continuum model (PCM) method was used to consider the implicit role of the solvent.62,63 Using the Solvation Model based on Density (SMD) solvation model, 64 the effect of solvation has also been calculated. In this model, the free energy of solvation is evaluated using the following equation
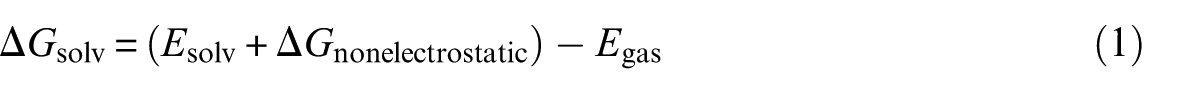
where Esolv and Egas represent the total energies of the system in the solution phase and gas phase, respectively, and
For the description of chemical reactivity and stability, quantum molecular descriptors such as hardness and electrophilicity index were used. The global hardness

where
Parr defined the electrophilicity index

Results and discussion
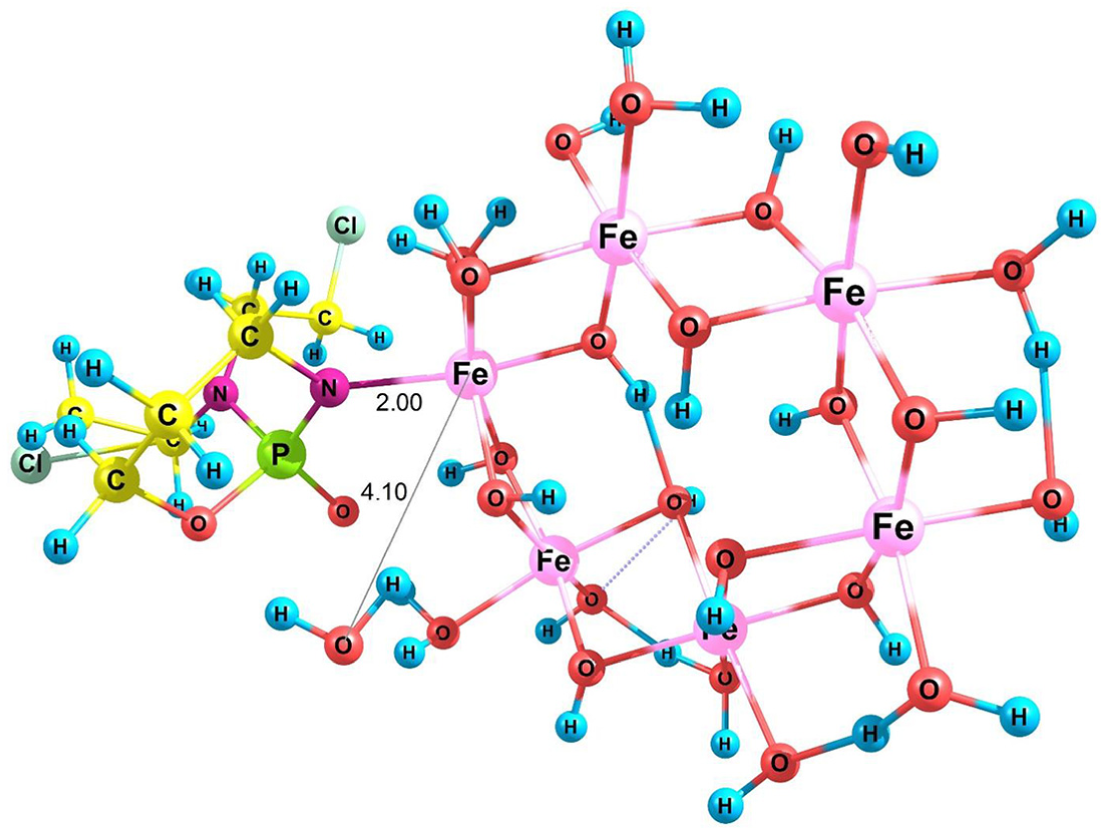
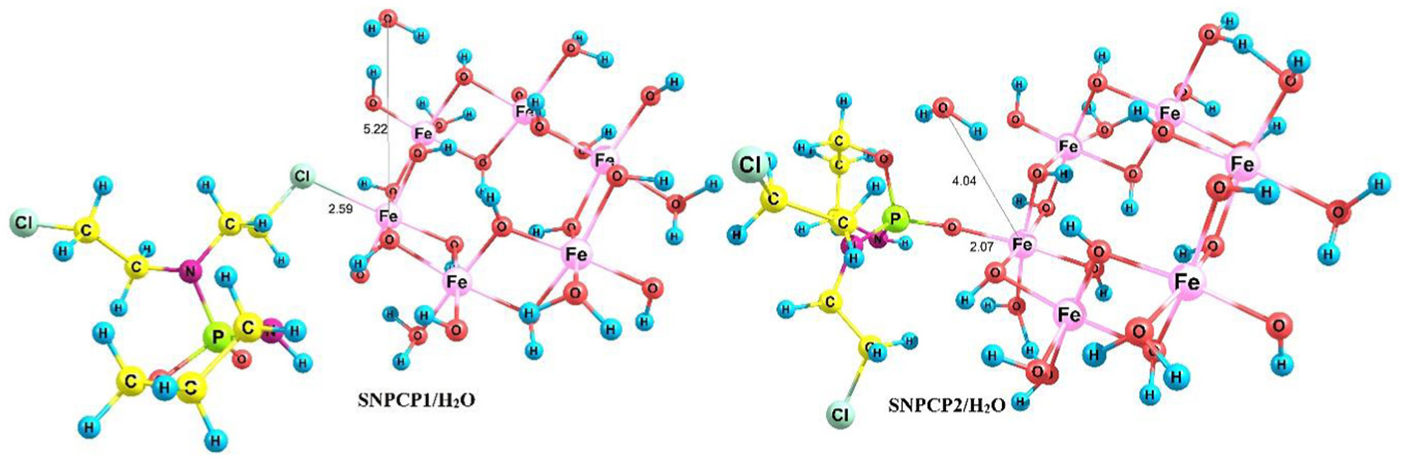
CP contains functional groups such as Cl, NH, and PO and has a nonplanar structure as shown in Figure 1. SNPs were modeled using γ-Fe2O3 nanoparticles in the solution phase (clusters of Fe6(OH)18(H2O)6 rings, including six-edge sharing octahedra joined via 12 OH groups and 6 surface OH and H2O 66 groups). The optimized geometries of the SNP γ-Fe2O3 and CP in the solution phase (water) are shown in Figure 1. Different configurations, during which the CP drug approaches the SNP via Cl, PO, and NH groups were called SNP/CP1, SNP/CP2, and SNP/CP3, respectively. For PO and NH groups, one structure was obtained (SNP/CP2,3). These reactants form hydrogen and pseudo-hydrogen bonds (Figure 2).
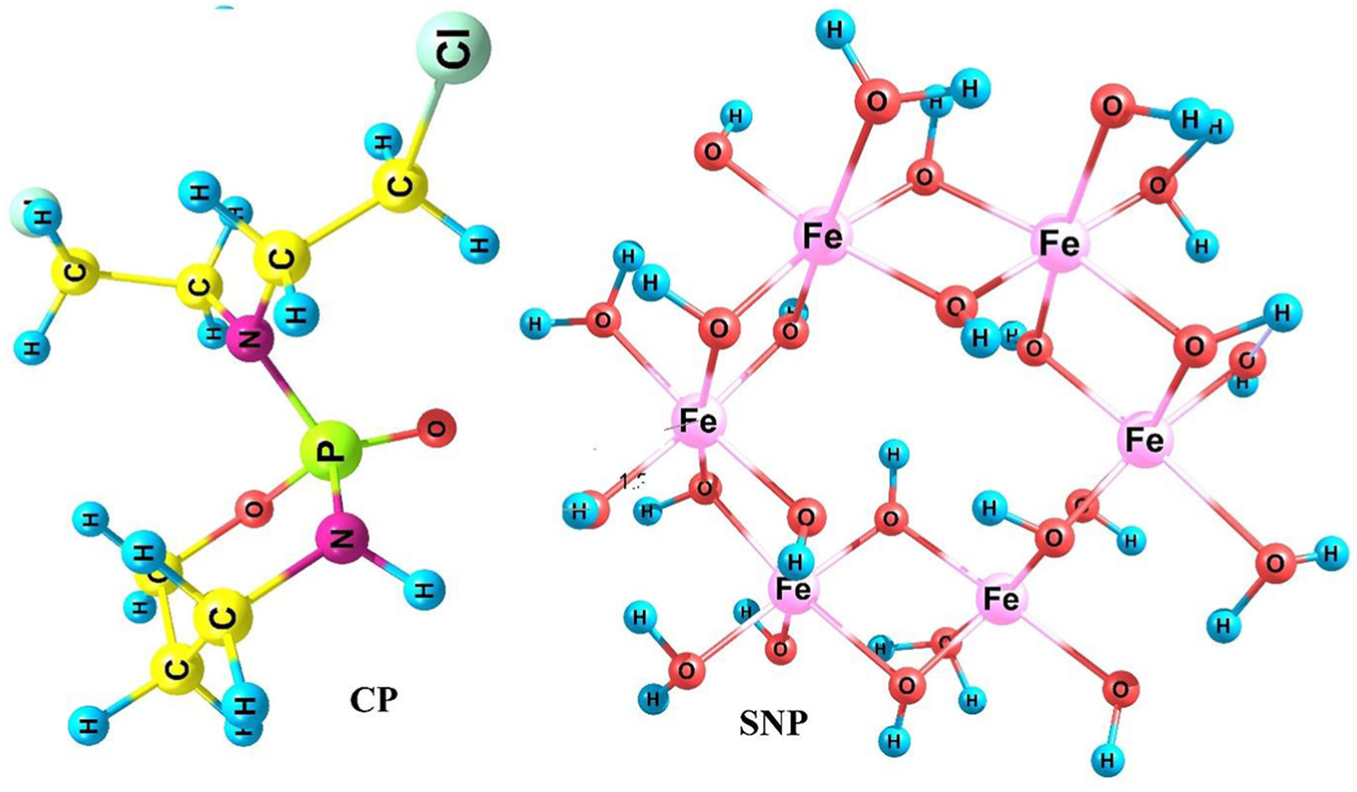
Optimized structures of SNP and CP.
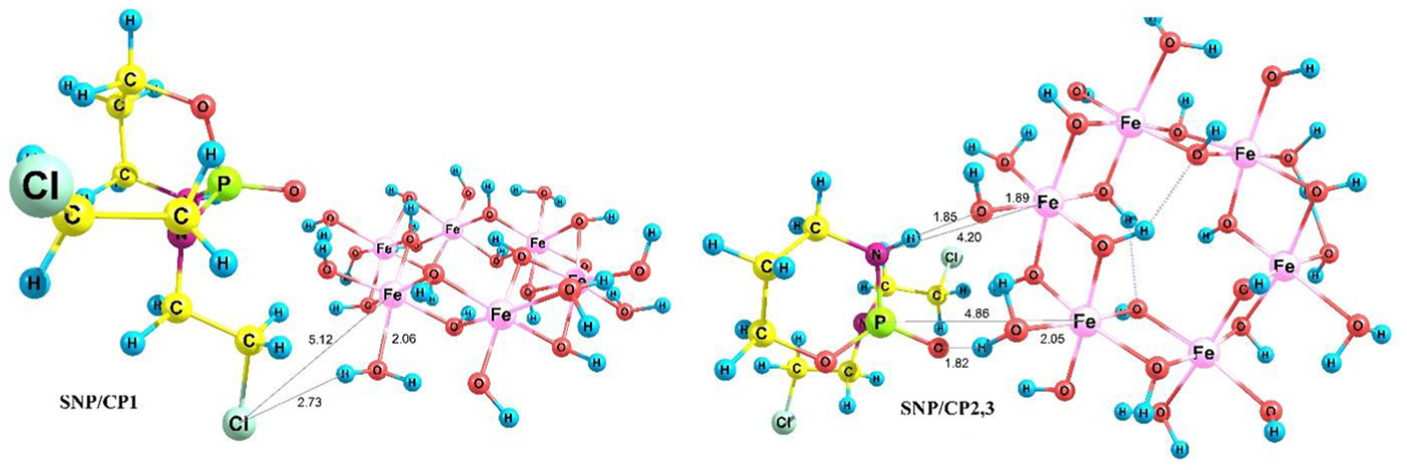
Optimized structures of SNP/CP1 and SNP/CP2,3.
The free energies of solvation
Solvation and binding energies of different configurations (kJ mol−1).
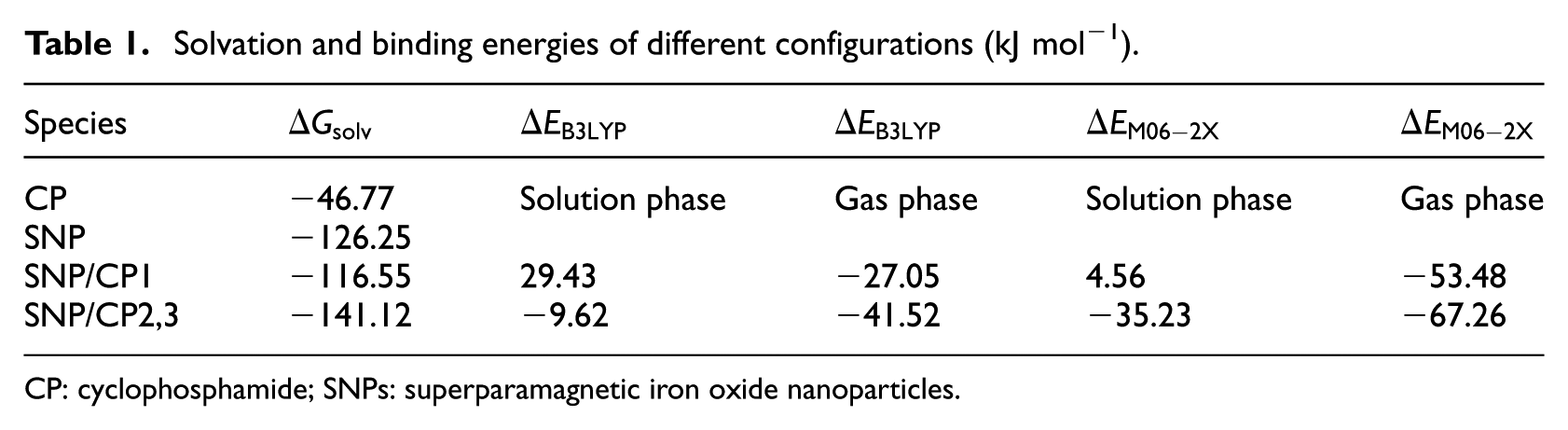
CP: cyclophosphamide; SNPs: superparamagnetic iron oxide nanoparticles.
The binding energies
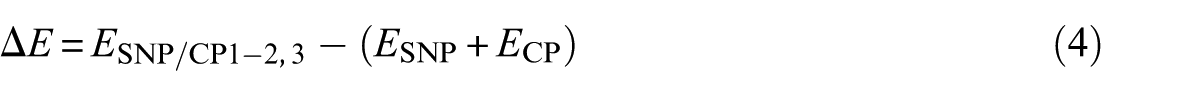
The calculated binding energies of SNP/CP2,3 are negative in both gas and solution phases. SNP/CP2,3 is more stable than SNP/CP1 in both phases. We repeated the calculations using the M06-2X functional to consider the contribution of dispersion interactions. The calculated binding energies related to M06-2X are more negative than those related to B3LYP (Table 1). Comparison between SNP/CP2,3 and SNP/CP1 shows that the former is more stable due to the stronger hydrogen bond between CP and SNP.
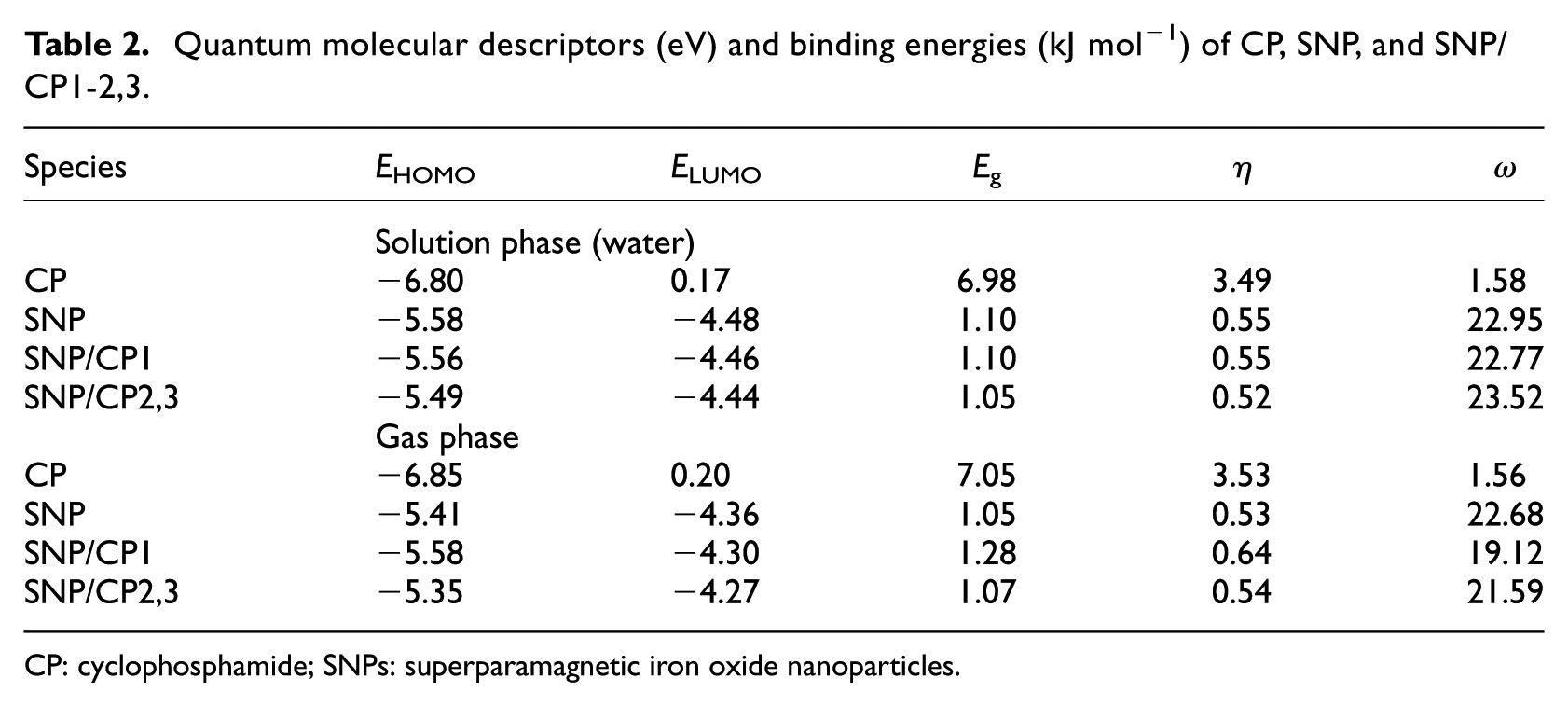
Quantum molecular descriptors for CP, SNP, and SNP/CP1-2,3 in both phases are represented in Table 2. In Table 2, Eg is the energy gap between LUMO and HOMO. Table 2 shows that the calculated values of η and Eg related to CP are remarkably more than those of SNP/CP1-2,3, meaning that the reactivity of the drug increases in the vicinity of the SNP. Also, ω for CP increases in the presence of the SNP, showing that CP acts as an electron acceptor.
Quantum molecular descriptors (eV) and binding energies (kJ mol−1) of CP, SNP, and SNP/CP1-2,3.
CP: cyclophosphamide; SNPs: superparamagnetic iron oxide nanoparticles.
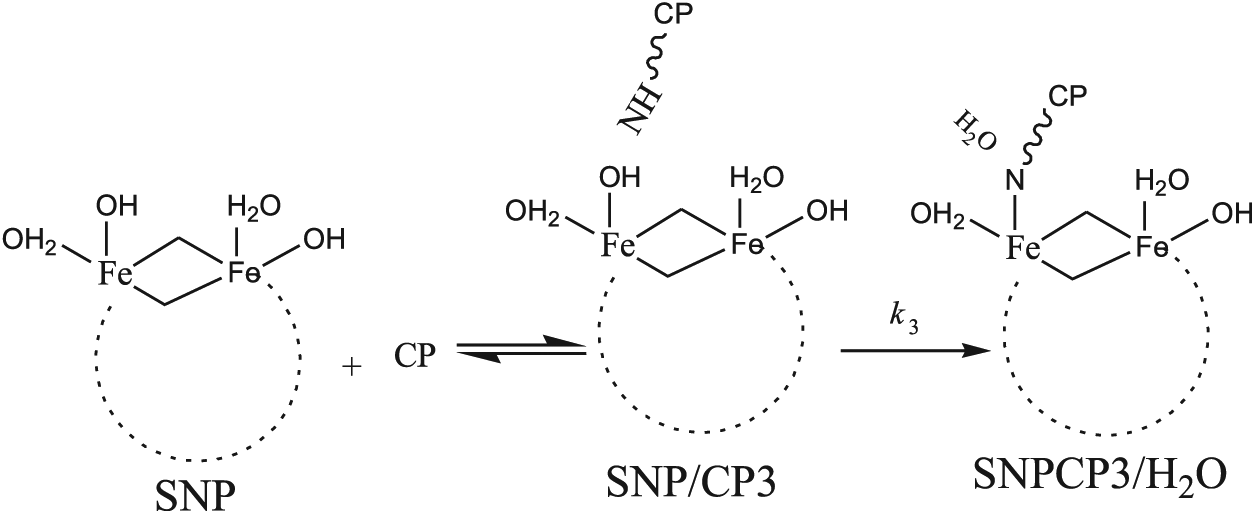
In order to study the possibility of formation of a covalent bond67,68 between the SNP and CP, we considered three mechanisms. First, we selected SNP/CP3 for the verification of covalent functionalization in the solution phase. In this case, NH in SNP/CP3 attacks the Fe atom to transfer its proton to the surface OH group of the SNP (Scheme 1). In this mechanism, reactant SNP/CP3 loses an H2O molecule to give product SNPCP3/H2O. The optimized structure of product SNPCP3/H2O is shown in Figure 3.
Mechanism k3.
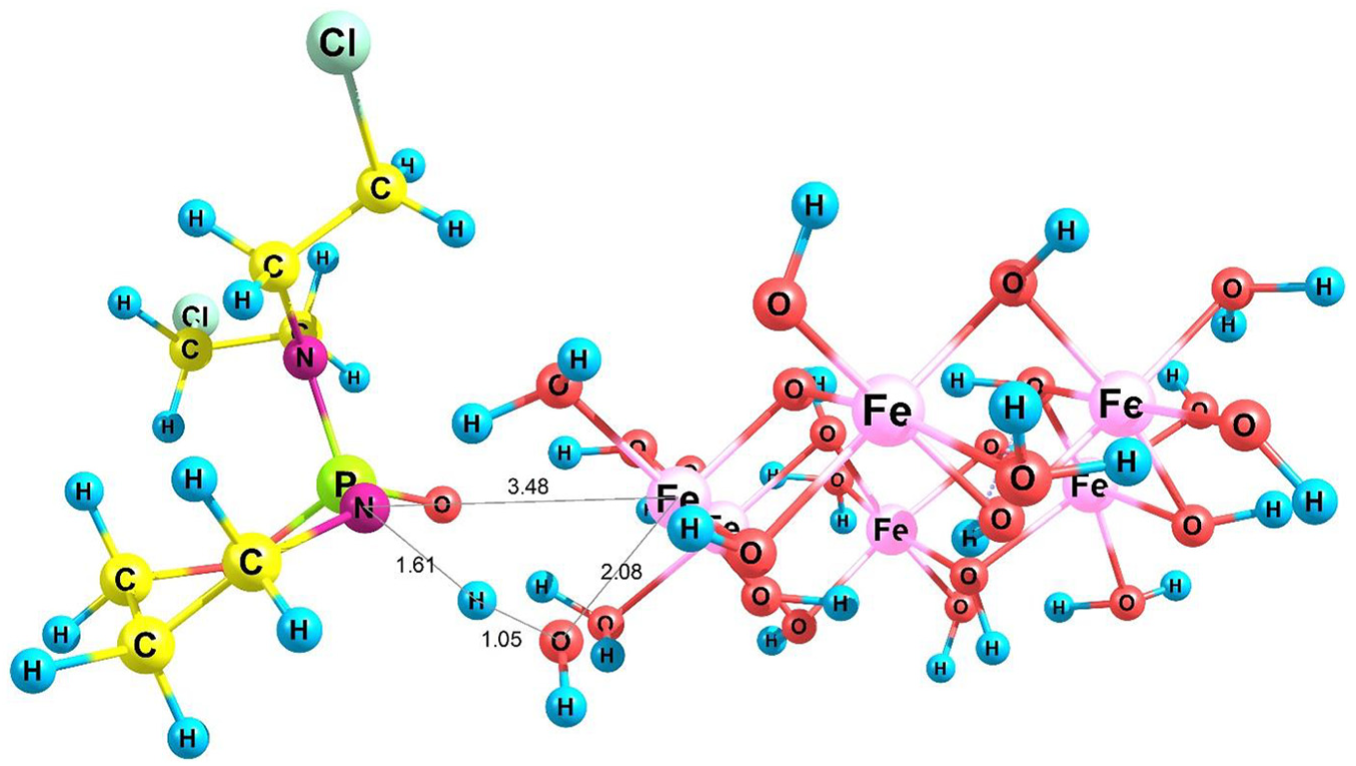
Optimized structure of SNPCP3/H2O.
The transition state of step k3 (TS k 3 ) was obtained through reactant SNP/CP2,3 and product SNPCP3/H2O and is presented in Figure 4. The calculated bond lengths for this mechanism are shown in Figures 2 –4.
Optimized structure of TS k 3 .
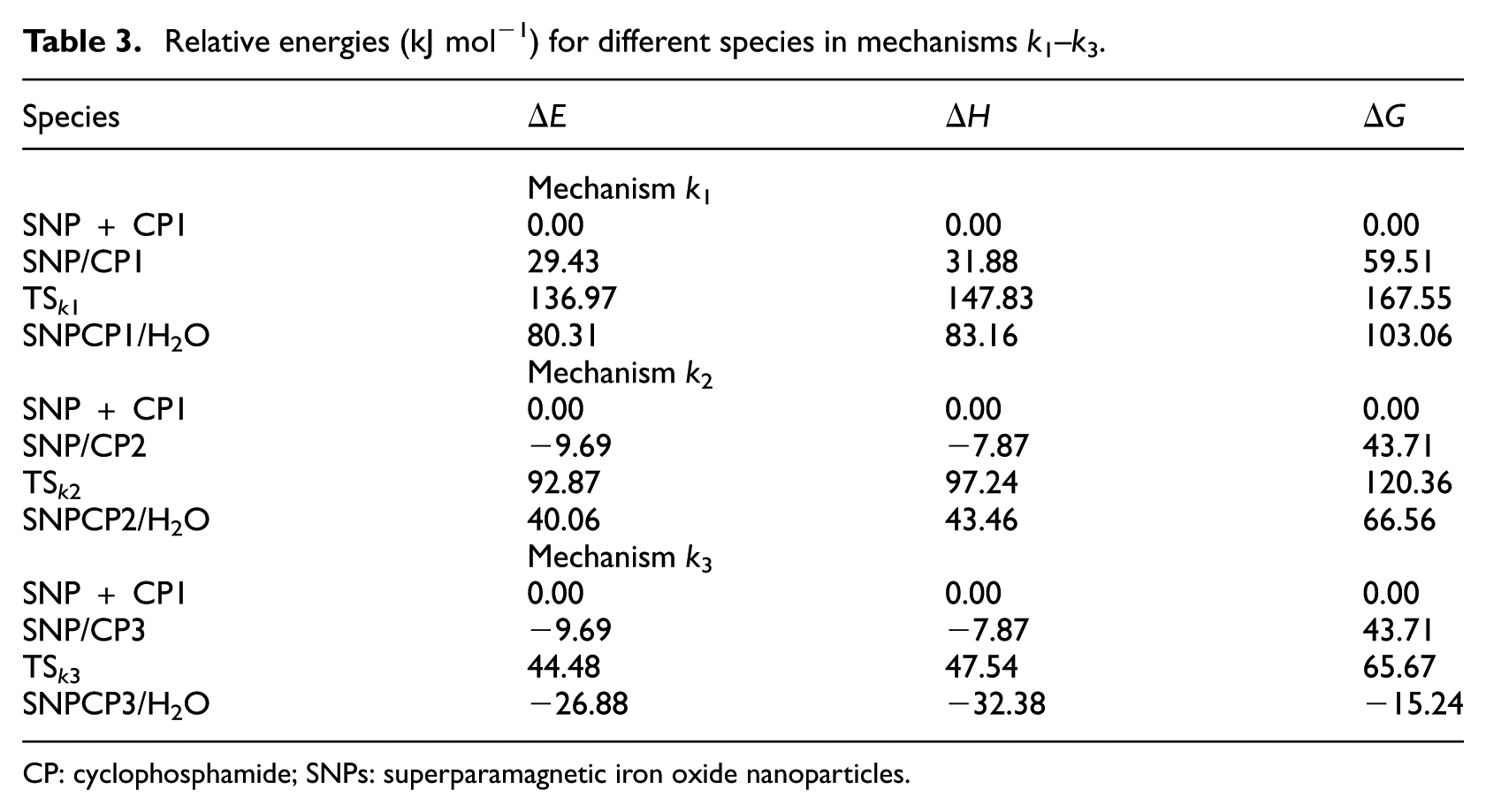
If the electronic plus zero-point energy (E), enthalpy (H), and Gibbs free energy (G) of SNP + CP become equal to zero, then the relative energies of the optimized structures in all pathways will be obtained. The total activation energy
Relative energies (kJ mol−1) for different species in mechanisms k1–k3.
CP: cyclophosphamide; SNPs: superparamagnetic iron oxide nanoparticles.
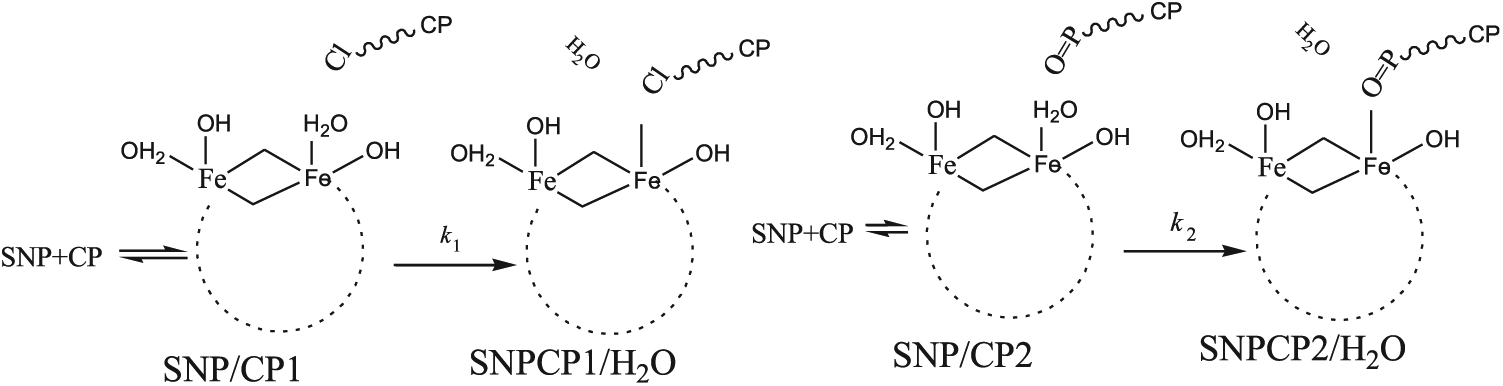
Scheme 2 demonstrates mechanisms k1 and k2 for the covalent functionalization of CP onto the SNP. In these reactions, the surface H2O of the SNP is substituted by the Cl and P=O groups of CP to give products SNPCP1-2/H2O (Figure 5).
Mechanisms k1 and k2.
Optimized structures of SNPCP1/H2O and SNPCP2/H2O.
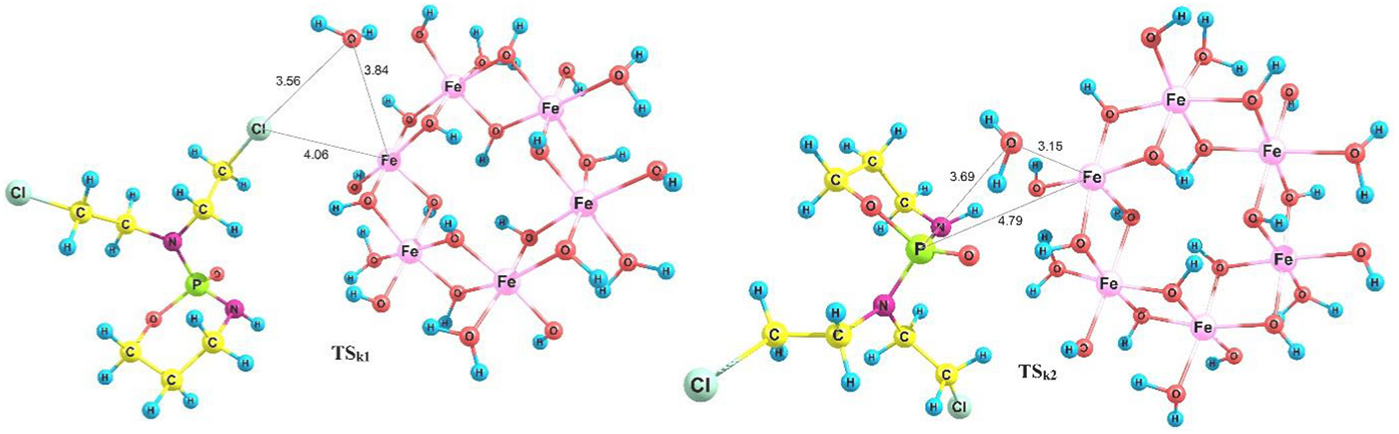
The transition states of step k1 (TS
k
1
) and step k2 (TS
k
2
) were optimized by using reactants SNP/CP1-2 and products SNPCP1-2/H2O (Figure 6). Using Table 3,
Optimized structure of TS k 1 and TS k 2 .
The activation energy for mechanism k3 is lower than for mechanisms k1 and k2 by 92.49 and 48.39 kJ mol−1, respectively. The product of pathway k3 (SNPCP3/H2O) is more stable than those of pathways k1 and k2 (SNPCP1-2/H2O). In contrast to the paths k1 and k2 which are endothermic and nonspontaneous, the reaction related to mechanism k3 is spontaneous (ΔG = −111.28 kJ mol−1) and exothermic (ΔH = −168.97 kJ mol–1). The energy barriers show that covalent functionalization can occur through NH in a six-membered ring. At room temperature, the activation energies related to mechanisms k1 and k2 are too high for covalent functionalization to take place through these pathways.
Conclusion
Two configurations of noncovalent functionalization of SNPs with CP were investigated at the B3LYP and M06-2X density functional levels in the gas and solution phases. The SNP was modeled using the Fe6(OH)18(H2O)6 ring cluster. The values of the solvation and binding energies show that these noncovalent interactions (SNP/CP) are energetically favorable. The values of global hardness and HOMO–LUMO energy gap related to CP are higher than those of SNP/CP1-2,3, showing the reactivity of CP increases in the vicinity of the SNP.
We studied three possible mechanisms of covalent functionalization of the SNP with CP thorough Cl (mechanism k1), PO (mechanism k2), and NH (mechanism k3) groups. The activation parameters related to mechanism k3 are lower than those of mechanisms k1 and k2. Pathway k3 is also spontaneous and exothermic. Therefore, our calculations show that mechanism k3 provides the possibility of creating a covalent bond between CP and SNP, while mechanisms k1 and k2 are not suitable for this task due to their high energy barriers.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received financial support for the research, authorship, and/or publication of this article: The authors thank the Research Center for Animal Development Applied Biology for allocation of computer time.