
Abstract
Evaluation of premarketing drug safety in clinical trials is often limited, due to the relatively small sample size and short follow-up time. The data collected in the postmarketing spontaneous reporting systems such as Food and Drug Administration Adverse Event Reporting System as well as electronic medical record systems provide crucial information to evaluate postmarketing drug safety. In this article, we assess the strengths and limitations of Food and Drug Administration Adverse Event Reporting System and electronic medical record data in studying the postmarketing pharmacovigilance outcomes for 12 selected antidepressant drugs. In addition, we evaluate the consistency of the results obtained from these two data sources, and provide potential directions for evidence integration.
Introduction
Mental illness is the leading cause of disability worldwide. 1 Statistics from the World Health Organization show that one in four people are affected by mental disorders. 2 In addition, mental health also impacts people’s physical health conditions. Although mental illness is more commonly seen than cancer or diabetes, less attention has been paid to it by investigators.
In the past decades, several structurally related and functionally similar drugs have entered the market for the treatment of mental disorders. Some of the antimanic drugs are chemically similar to existing drugs without evidence of treatment improvement.3,4 Studies have been conducted to compare the efficacy and acceptability of mental health drugs through randomized controlled clinical trials and meta-analyses.5–7 Premarketing clinical trials are also standard methods for detecting adverse drug reactions before releasing drugs into the market. However, some adverse events may be unanticipated and could occur after the Food and Drug Administration (FDA) approval of the drug. Hence, postmarketing surveillance analysis remains critical. 8
Spontaneous reporting systems (SRSs) of discovered adverse reaction play an important role as part of the postmarketing surveillance. For example, the US FDA has initiated an SRS, known as the FDA Adverse Event Reporting System (FAERS), which contains over 6,156,081 reports between 2004 and 2015 provided by health providers, patients themselves, and so on. 9 We have previously investigated this database to explore the feasibility of using FAERS to analyze mental health drug safety using 12 first-line antidepressants drugs. 10 We found that our results were qualitatively in agreement with results from the recent comprehensive systematic review of randomized controlled trials. 5 Moreover, our postmarketing analysis based on FAERS provided more insight into different types of adverse reactions with respect to different classes of human organ system and a larger population. These findings served as an important evidence for drug safety, and could compliment premarketing clinical trials to better aid clinical decision-making.
However, limitations do exist for SRS. First, unlike clinical trials, the adverse events from SRS do not have a fixed time frame between medication and event for each patient. 9 Therefore, the temporal relationship between the drugs and the reported adverse events is not clearly specified. Also, patients or health providers only report if an adverse reaction arose, such that data of the control group, that is patients taking the same medication who did not experience adverse reactions, are not available. Furthermore, whether an adverse event will be reported or not depends on many factors, such as age, gender and race, 11 leading to unbalanced distributions of variables for statistical analysis.
Some of these disadvantages of SRS can be remedied by leveraging the power of electronic medical records (EMRs). EMRs contain more detailed time-related information on patients’ encounters with health systems, such as the admission and discharge dates for patients’ encounters, and the start and end dates for medication orders. Such information provides opportunities for studies using temporal information as a factor to investigate relations between adverse reactions and prescribed medications. Also, EMR data are collected through routine clinical care. Much more details for patient visits were recorded in the EMR system compared to the SRS. Nevertheless, certain limitations still exist for using EMR data for drug safety analyses. For example, since SRSs are designed to study the adverse events, they are written in the format of standardized terminology for adverse reactions named Medical Dictionary for Regulatory Activities (MedDRA). 12 In contrast, EMRs usually use standard codes such as the International Classification of Diseases (ICD) codes for diagnosis results, which cannot be directly used as a representation of adverse reaction.
To overcome the limitations of a single resource (e.g. SRS) for studying drug safety, researchers have made efforts to integrate multiple data sources to enhance the quality of drug safety surveillance. Li et al. 11 investigated whether combining SRS with electronic health records (EHRs) could improve the performance of adverse reaction detection. Harpaz et al. 13 proposed a signal-detection method combining the SRS and EHRs by requiring signals from both sources. Xu and Wang 14 combined biomedical literature and FAERS in order to improve postmarketing drug safety signal detection. However, these studies only used the drug name and adverse reactions generated from each data source and combined only the drug–adverse reaction pairs afterward. In our study, we preserved the initial information, such as the time relationship and data format for patient information, including encounter times and gender, by mapping the diagnosis code directly to the standardized adverse reaction vocabularies. The direct mapping is more flexible than combining SRS and EMR for analysis.
Specifically, in this paper, we evaluated the evidence consistency of pharmacovigilance outcomes between FAERS and EMR data for acute mania patients. We considered safety outcomes of 12 first-line antimanic drugs: aripiprazole, carbamazepine, valproate, gabapentin, haloperidol, lamotrigine, lithium, olanzapine, quetiapine, risperidone, topiramate and ziprasidone from FAERS, and compared the safety signal in EMR data by applying a data mining method in a large-scaled EMR system and extracting encounters and medication orders status for 772,181 unique patients. Since the EMR system implemented ICD as a description for the state of an illness of patients, we mapped ICD to MedDRA so that our comparison of evidence is on the same terminology. We then quantified the consistency of evidence from two data sources statistically.
Method
Data source
We considered two data sources in this study. One was the Cerner Health Facts EMR database, and the other was a SRS, the FAERS. The EMR database can compliment to FAERS and overcome some of its limitations, for example, lack of control group and temporal relationship. The detailed comparison of two data sources is shown in Table 1.
Comparison of two data sources.
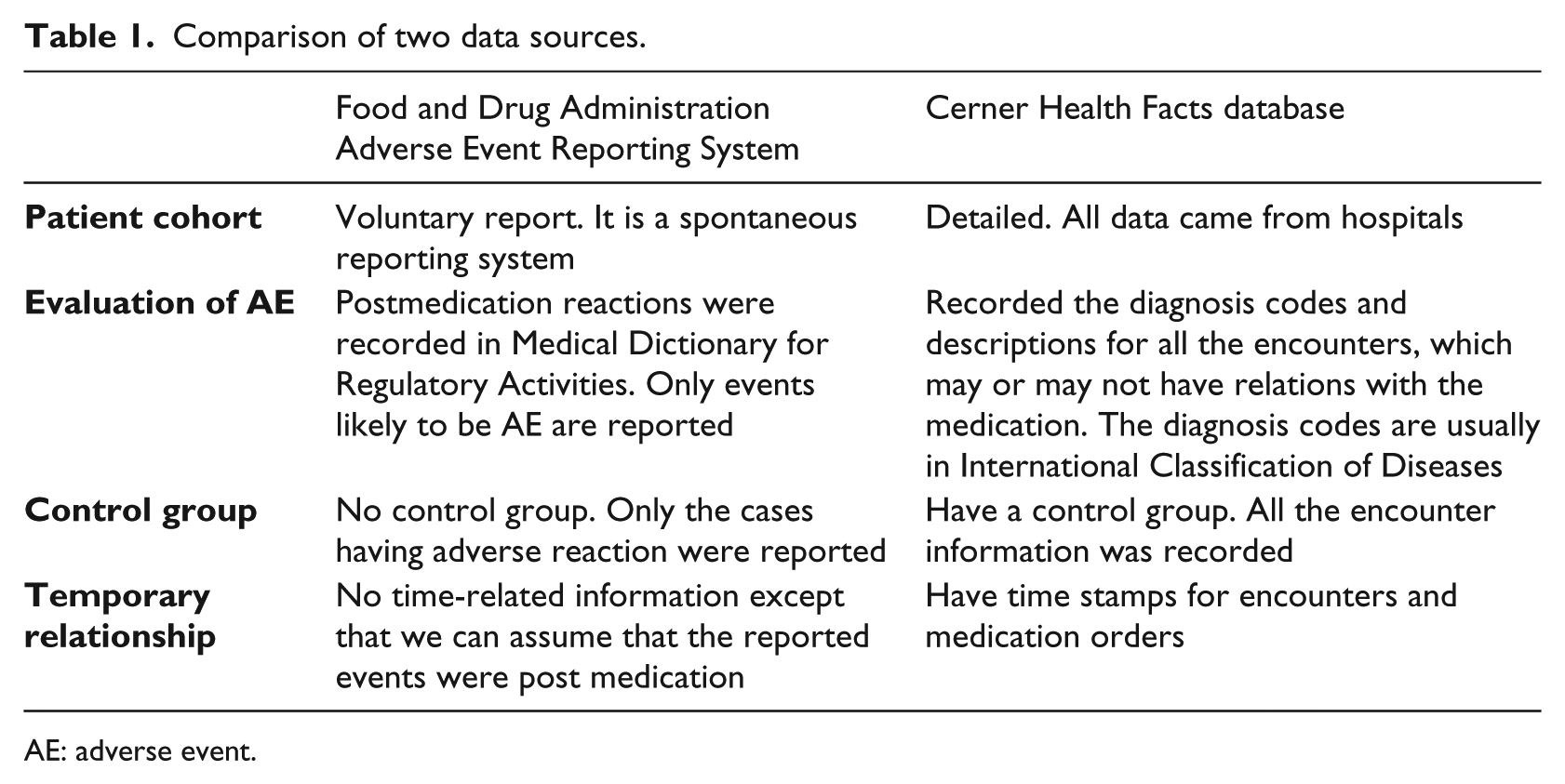
AE: adverse event.
Cerner Health Facts EMR database
Cerner Health Facts EMR database is a Health Insurance Portability and Accountability Act (HIPAA)-compliant database collected from several enrolled clinical facilities, containing mostly in-patient data. Data in Health Facts is extracted directly from the EMR from hospitals in which Cerner has a data use agreement. Encounters may include the pharmacy, clinical and microbiology laboratory, admission and billing information from affiliated patient care locations. All admissions, medication orders and dispensing, laboratory orders and specimens are date- and time stamped, providing a temporal relationship between treatment patterns and clinical information. Cerner Corporation has established operating policies consistent with the HIPAA Laws to establish de-identification for health facts. It contains clinical records with around 1.3 billion laboratory results, 84 million acute admissions, emergency and ambulatory visits and 9 years of detailed pharmacy, laboratory, billing and registration data as well as 151 million orders for medications from all patients along with time-stamps and sequences information. 15
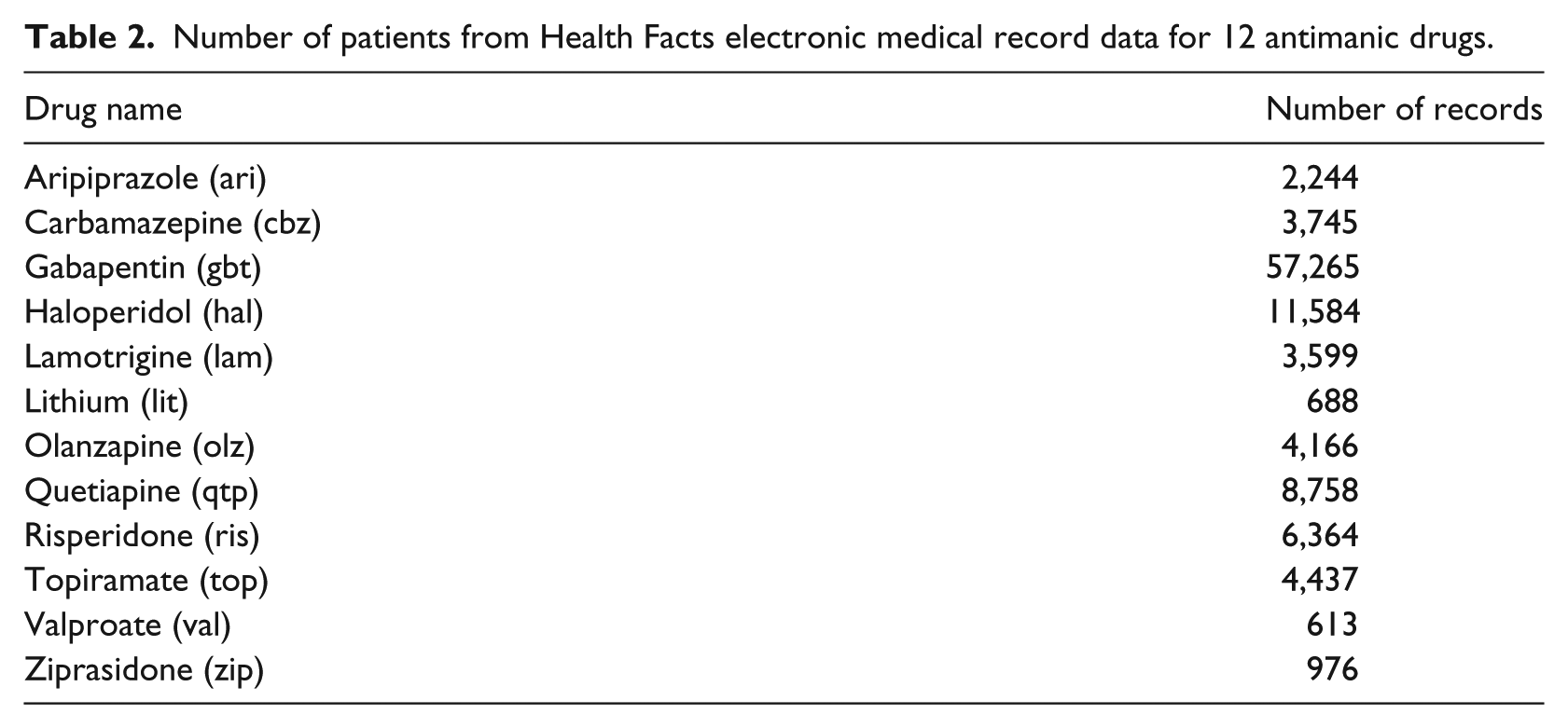
We obtained 772,181 patients from the Cerner Health Facts database who were prescribed at least one of the 12 antimantic medications. As some of the patients were on and off different drugs, it was hard to determine which drug caused a certain adverse event. Therefore, we restricted our study population to patients who were prescribed only one of the 12 drugs. To avoid irrelevant diagnosis codes, we removed all the diagnosis codes that had been entered in the last encounter before the start date of the medication. Since the patients we extracted from Health Facts EMR data were all inpatients, there were two time stamps corresponding to each encounter: the admission time and discharge time. We then defined a selection window from the start date of medication and 180 days after the end date of the medication. All postmedication diagnosis codes with the admission and discharge dates within the selection window were selected (Figure 1). Patients without such event were eliminated. Our final study population includes 104,439 patients. Figure 2 shows the number of patients removed in each selection step. Table 2 shows the number of patients selected for each drug.
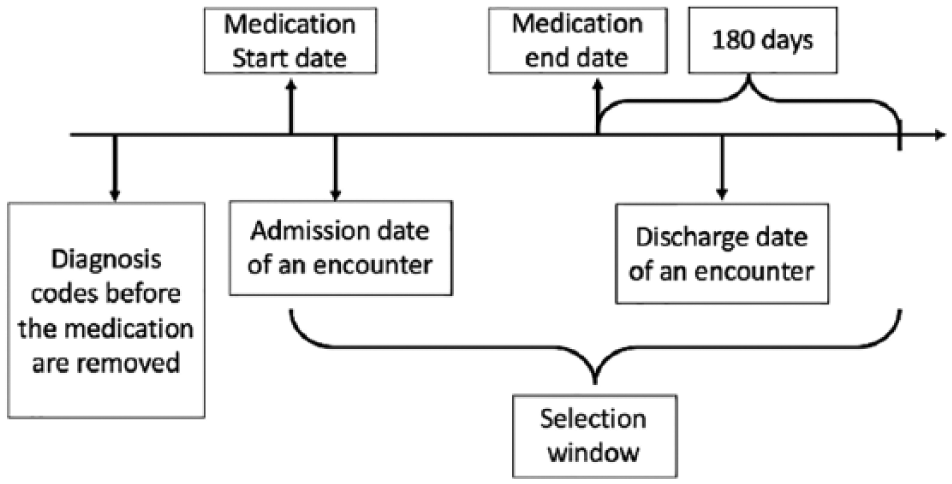
Electronic medical records cohort identification based on medication and encounter dates.
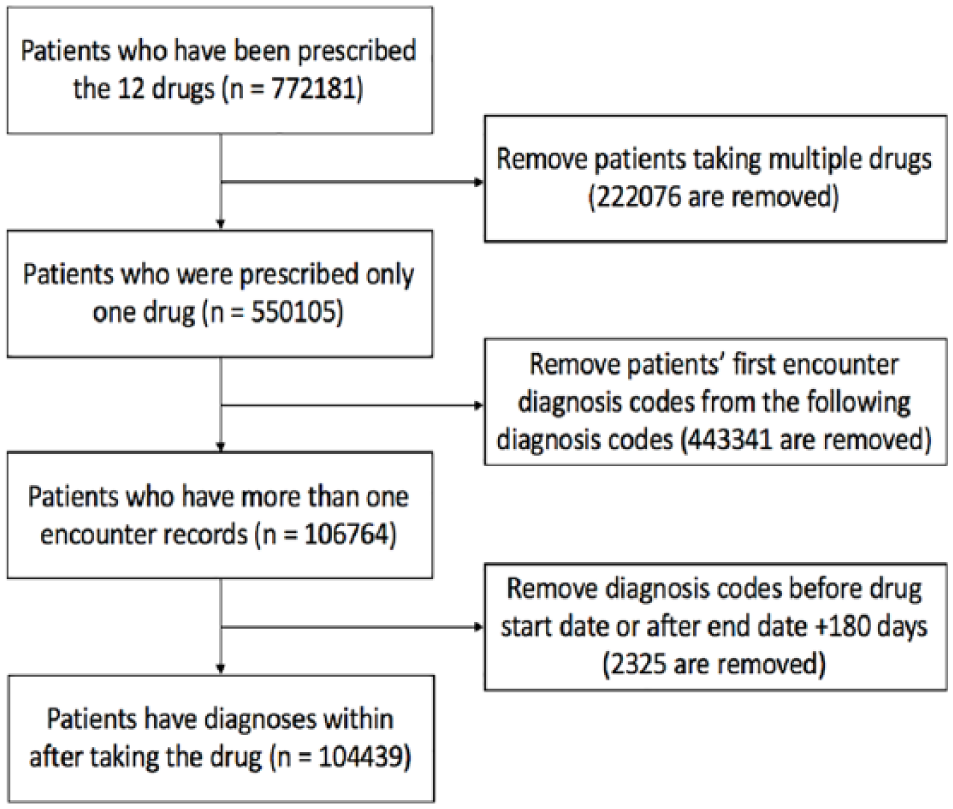
Number of patients who remained after electronic medical record cohort identification.
Number of patients from Health Facts electronic medical record data for 12 antimanic drugs.
FAERS
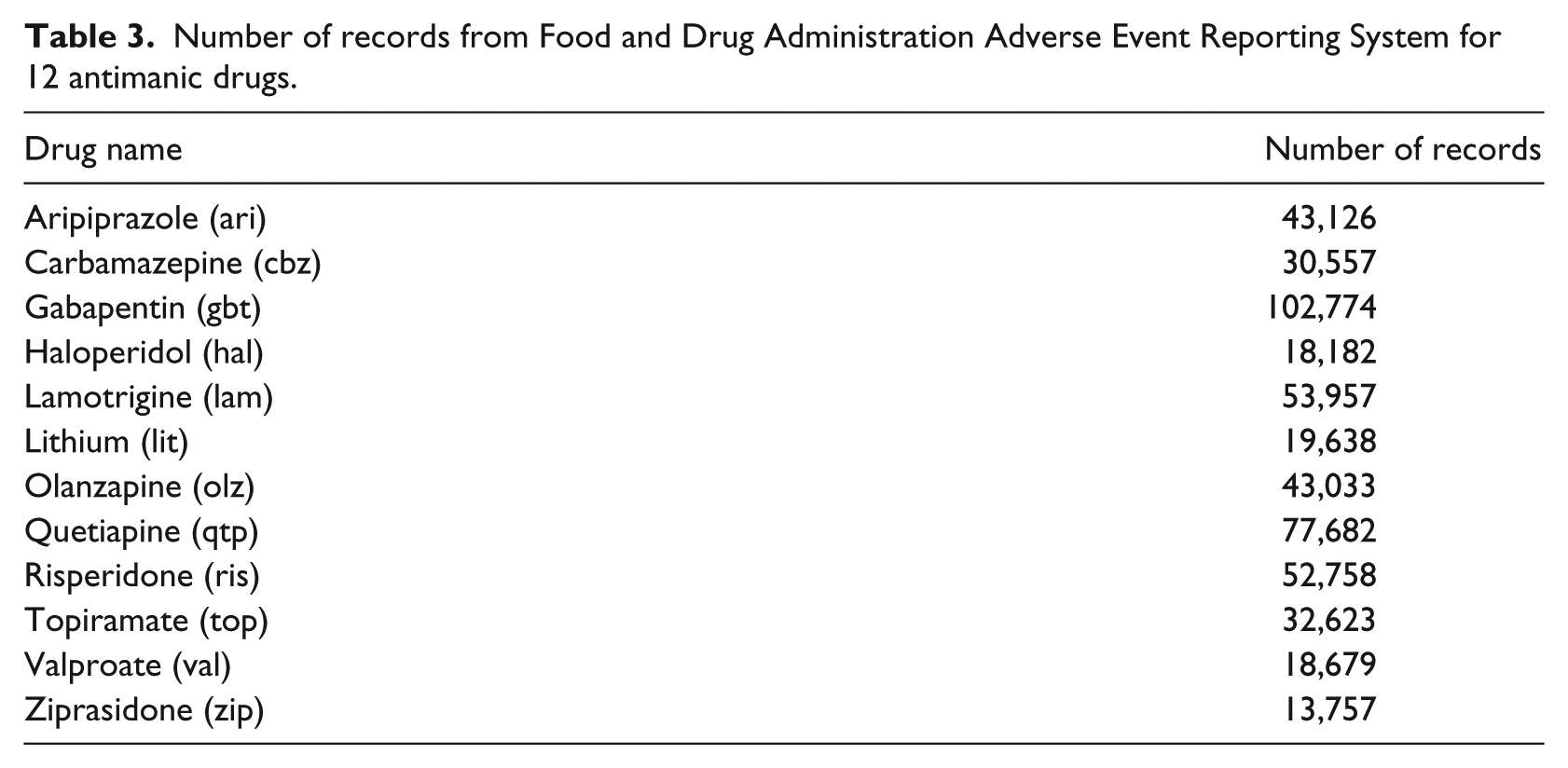
FAERS contains 6,156,081 reports from the past 13 years. It is an SRS where the reports submitted are evaluated by the Center for Drug Evaluation and Research and the Center for Biologics Evaluation and Research. 8 We extracted data for the 12 antimanic drugs in FAERS from 1 January 2004 to 31 December 2015. Details are shown in Table 3.
Number of records from Food and Drug Administration Adverse Event Reporting System for 12 antimanic drugs.
Data extraction method
We searched the Health Facts EMR database by brand and generic names of 12 antimanic drugs, and generated two tables. One of them consisted of the unique patient identification number, encounter identification number, medication start and end dates, diagnosis code, age and gender. The other contains all the encounter IDs from the first table, along with the encounter time stamps and diagnosis type. According to the definition provided by Cerner Health Facts, 12 most of the diagnosis types equate to the final diagnosis for the visit. So we only considered the diagnosis code that was given at discharge for each encounter.
Terminology mapping
We used the common data model from the Observational Health Data Sciences and Informatics (OHDSI) to generate an overall table for all the ICD, 9th Revision, Clinical Modification codes mapping to MedDRA. The common data model preserved the relationships between various terminologies.
The MedDRA terms from FAERS are on the “PT” level, referring to “preferred term” in MedDRA. MedDRA provides a hierarchical structure of the terms where PT is the second lowest level descriptor for adverse events. Hence, we mapped all the levels of MedDRA terms in the generated table to “PT” level referring to the MedDRA data table. Then we joined this table with the extracted Cerner Health Facts table with the limitation that all the PT terms had to be within the range of the previous study of FAERS. Finally, we generated a table where the encounter and medication data were linked with “PT.” Some information was lost due to mapping coverage. Figure 3 shows the workflow of this terminology mapping step.
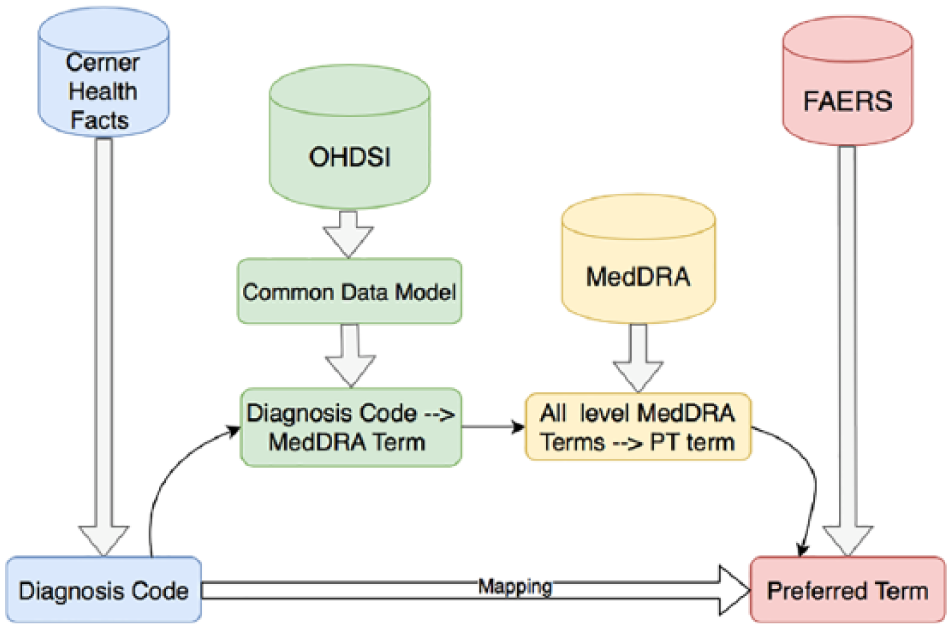
Terminology mapping from Cerner Health Facts database to FAERS using common data model in OHDSI.
Statistical method
With the patient records extracted from the Cerner Health Facts database, we carefully defined the adverse events, and studied the top adverse events ranked by their prevalence. We quantified the agreement of top adverse events identified by EMR data and FAERS data. We also fitted generalized-linear regression models for the total number of adverse events identified or reported by each patient adjusted by age and gender, and compared the results obtained from EMR with the results from FAERS data. Using EMR data, we studied the dropout rate for each drug and compared it with the premarketing clinical trials.
Identifying the top adverse events
We calculated the prevalence of adverse events for each drug, and compared the top 100 adverse events identified by FAERS and EMR data. We obtained the adverse events identified by both databases, where the number of overlapping adverse events can be considered as a measure of agreement of the evidence from these two data sources.
Modeling the number of adverse events per patient
For each patient, we summarized the total number of adverse events reported. Since a patient had to have at least one report to be included in the dataset, the total number of adverse events for each patient is a positive integer. To account for potential overdispersion, we modeled the total number of adverse events using a zero-truncated negative binomial distribution. This model assumes the total number of adverse events for each patient follows a zero-truncated negative binomial distribution with the following probability mass function:

where
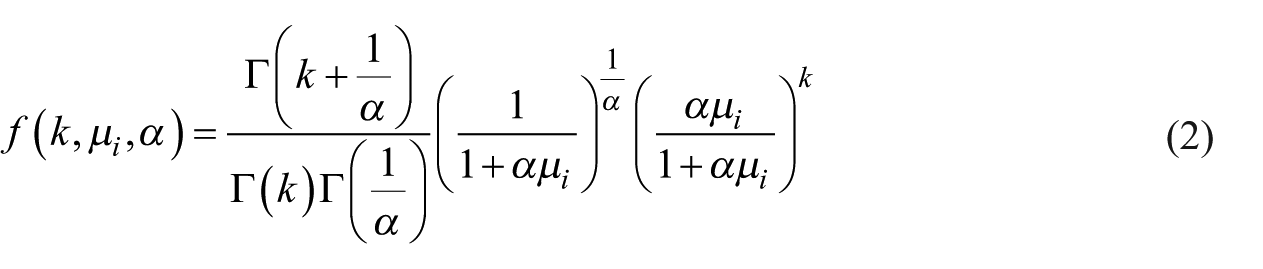
In equation (2), α is the overdispersion parameter and µi is the mean of the negative binomial distribution, that is, event rate (or expected counts) of having a particular adverse event. In this study, the overdispersion parameters were set to be equal for all the drugs since we did not observe large variation of
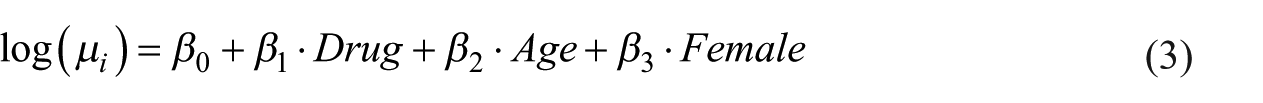
In both datasets, the 12 antimanic drugs were coded as a categorical independent variable with 12 levels, and aripiprazole is set as the reference group. Therefore, we obtained estimated age- and gender-adjusted log ratios of the expected counts for all other 11 drugs compared to aripiprazole. Then the regression was fitted using R package “VGAM” and the log rate ratios were estimated with 95 percent confidence intervals (CIs). The drugs were then ranked based on the rate ratios.
Ranking the dropout rate
Among the 222,076 patients who were prescribed multiple drugs, we defined a dropout for a drug if a patient switched from this drug to another drug, which is an important measure of the overall lack of efficacy/safety. The dropout rate was calculated from the ratio of the number of dropouts of a drug to the number of patients who were prescribed the drug. We calculated the dropout rates of each drug and compared them with the dropout rates reported in a meta-analysis of premarketing clinical trials.
Results
The average age of the final selected patients in EMR was 64.09 (95 percent CI: 63.98, 64.21) and 42.24 percent of the patients were females. The average age of the reports in FAERS was 47.22 (95% CI: 47.19, 47.25) and 40.49 percent of the patients were females.
Table 4 shows the overlapping top 100 postmedication events. The largest number of overlapping adverse events is 11 for quetiapine, and there are no overlapping top 100 events for valproate.
Overlapped 100 adverse events from Food and Drug Administration Adverse Event Reporting System and Cerner Health Facts database.

Figure 4 shows the model-fitting results for the two data sources. Setting aripiprazole as the reference drug, the black dots in the plot were the point estimates of rate ratios of the 11 drugs compared to aripiprazole. The corresponding green line segments showed the 95 percent CIs. The ranking of the 12 drugs was generally not consistent between the two data sources. After controlling for age and gender, EMR data suggested that haloperidol had the smallest number of events per patient, and FAERS data suggested that risperidone had the smallest number of events per patient. On the other hand, gabapentin was suggested by the EMR data and carbamazepine was suggested by the FAERS data to have the largest number of events per patient.
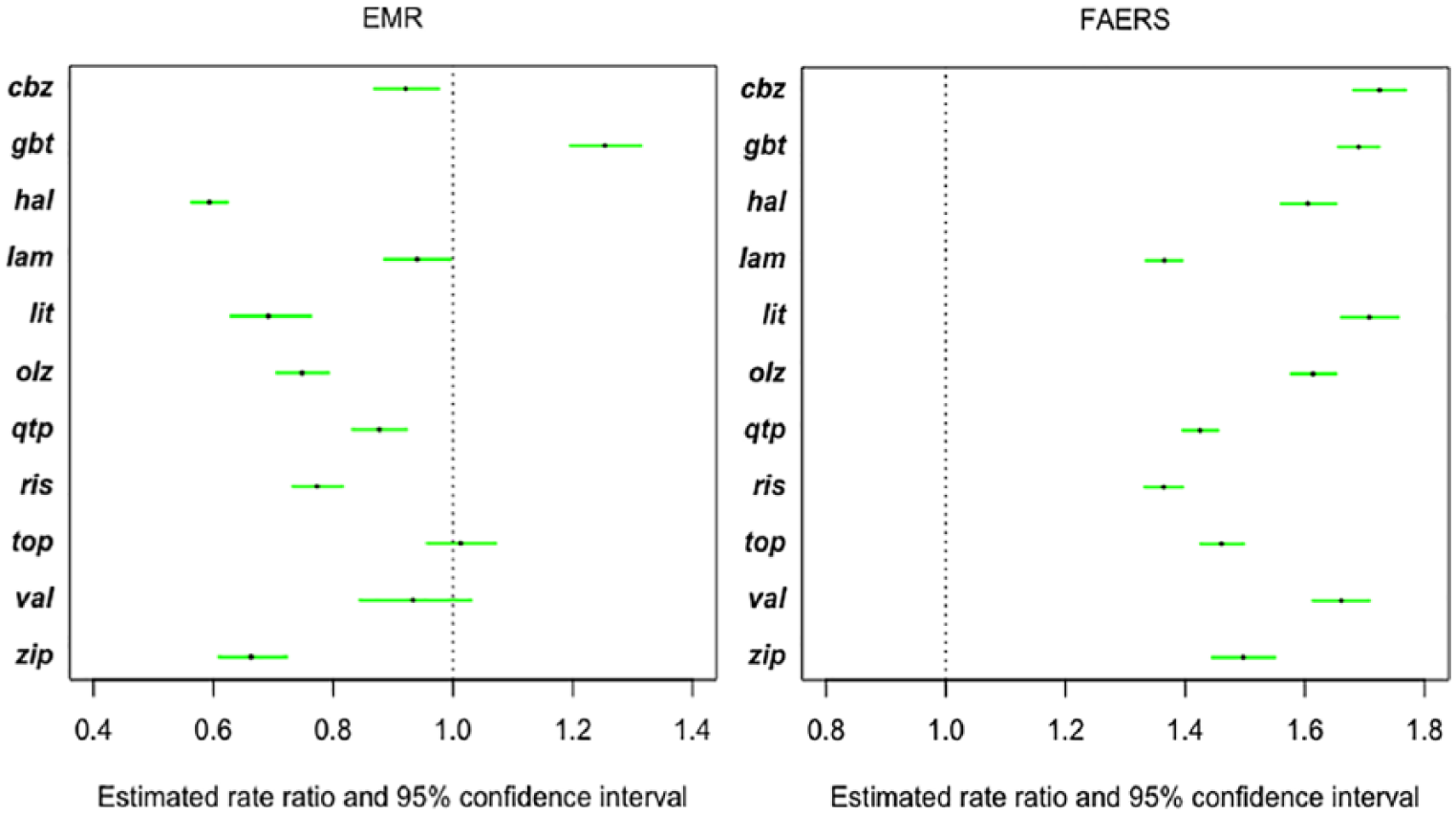
Estimated rate ratios and 95 percent confidence intervals from zero-truncated negative binomial model using EMR and FAERS data.
Using the dropout defined in the previous section, we plotted the dropout rates of the 12 drugs in Figure 5. The result showed that risperidone had the lowest dropout rate, where in the premarking meta-analysis, risperidone was showed to be the second highest drug ranked by acceptability. Ziprasidone, however, was suggested by the EMR data to be the drug with highest dropout rate, while it was ranked in the middle by the premarking meta-analysis. 4 However, the differences in the absolute dropout rates of the 12 drugs were small, and the small differences might have led to less power in correctly ranking the drugs. More importantly, the definition of dropout rate in EMR was different from the dropout rate in a clinical trial. The switch between medications may have multiple causes, including lack of drug efficacy, safety and other socioeconomic factors.
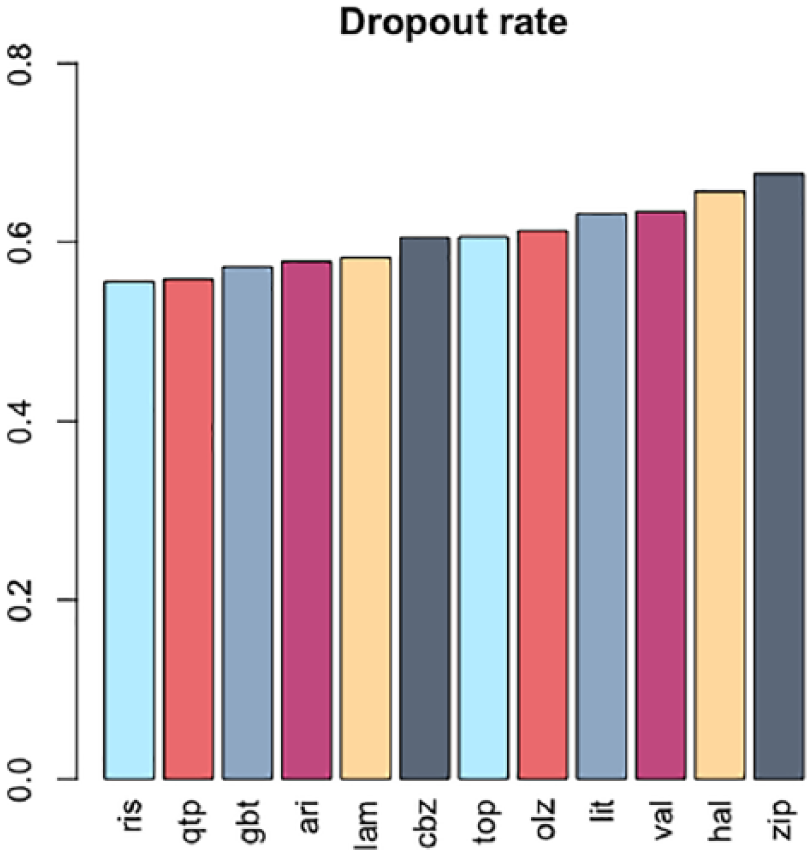
Dropout rates calculated using EMR data.
Discussion
The premarketing safety profile of medications is often limited because of the relatively small number of individuals included in the trials. Recently, there has been increasing interest in using data from postmarketing surveillance systems and medical records to study drug safety and efficacy in “real-world” settings. In this study, we performed drug safety analyses on 12 antimanic drugs using data from FAERS and the Cerner Health Facts database, and evaluated the feasibility of combining evidence from these two data sources. Synthesizing the evidence on drug safety from the FAERS database and the Cerner Health Facts database encountered a few key challenges in informatics and statistical analyses.
Using antimanic drugs as a use case study, we conducted a rigorous investigation by mapping safety terms to a common data model developed by the OHDSI consortium, carefully defined the patient cohorts, and compared the safety signals from two data sources through formal hypothesis testing procedures. We found that the top adverse events identified from each data sources only overlapped by a relatively small percent. By modeling the number of adverse events using a zero-truncated negative binomial model, the rankings of log rate ratios were generally not consistent between the two data sources. The dropout rates calculated by the Cerner Health Facts database do not agree much with the premarketing clinical trials either.
Although the difference in the population characteristics might lead to heterogeneity to certain degree, the failure of finding consistency between the evidence from different data sources reveals the important challenges of using EMR data for drug safety evaluation. From our investigation, we believe that correct identification of adverse events is the major difficulty in using EMRs to study drug safety. For instance, EMRs contain all encounter data for each patient. Some encounters may not be relevant for mental disorders or the specific drugs at all. In addition, patients who took the drugs that treat serious mental disorders are at increased risk of certain events such as suicide-related death. In this case, we cannot simply treat these events as adverse reactions to that drug. And if patients were taking multiple medications at the same time, it becomes challenging to understand which drug caused the adverse event. Despite these challenges, the promise of EMR data remains, especially due to the fact that the extremely large sample size in EMR data allows us to set rigorous criteria to exclude ambiguous records, and rare events could be ruled out by more sophisticated statistical modeling. Moreover, extracting controls can help distinguish adverse events from other types of diagnoses. However, the control group should be chosen carefully to match the case group to avoid confounding factors. Conclusions should be made with cautions and potential biases should be considered thoroughly.
Challenges also exist in using the FAERS data. Since FAERS is a passive reporting system, the medication information is more limited, in particular we do not know whether patients are taking multiple drugs unless they were reported. Moreover, the same adverse events might be reported multiple times for different drugs for the same patient, and it could reduce the power of statistical comparisons. Also, events are often reported without clear time stamps, and potential confounding factors are not included. As discussed in the literature, FAERS can be considered as a great source for signal detection, rather than validation. With safety signals detected in FAERS data, a replication study could be performed in the EMR or other types of data in order to draw more reliable conclusions.
Despite these challenges, the promise of EMR data remains, especially due to the fact that the extremely large sample size in EMR data allows us to set rigorous criteria to exclude ambiguous records, and rare events could be ruled out by more sophisticated statistical modeling. In addition, extracting controls can help distinguish adverse events from other types of diagnoses. Statistical methods should be further developed to properly match control patients with the patients under exposure, in order to achieve more concrete determination of adverse event from EMR system. Moreover, as the majority of health-related information is in narrative format, the clinical notes could contain more information regarding patient himself, for example, patient history. We could use various Natural Language Processing (NLP) techniques such as name entity recognition and (temporal) relation extraction to extract the mention of adverse reactions from clinical notes. Since ICD code was designed for billing purpose instead of adverse reactions reporting, extraction of adverse reactions from clinical notes might reveal more evidence.
In the future, informatics tools should be developed to extract information from both structured and unstructured data, and statistical methods should be further developed to properly match control patients with the patients under exposure, in order to achieve more concrete determination of adverse event from EMR system. And methods should be developed to better account for the selection bias, population heterogeneity and other challenges when combining EMR with FAERS data to study the postmarketing drug safety.
Conclusion and future direction
In this article, we showed that FAERS data and EMR both have strength and limitations in studying the postmarketing pharmacovigilance. FAERS data are clear in the definition of adverse events but lack temporal information and confounding measurements. EMR data have well-structured temporal information and covariates measures but are difficult as a source to identify adverse events and to define patients with exposure. To evaluate the evidence consistency of pharmacovigilance outcomes between FAERS and EMR data, we performed rigorous drug safety analyses for 12 first-line antimanic drugs. Importantly, the results from the two data sources reached low consistency. This finding revealed a few key challenges in using multiple resources for drug safety analyses, including proper definition of adverse events under different contexts. More sophisticated informatics and statistical modeling tools need to be developed to bridge these gaps for evidence synthesis. In the future, we plan to include data extracted from clinical notes, which potentially would cover additional information directly relevant to adverse events. We will also explore more systematic ways to define the control groups using EMR data, and more robust evidence synthesis models.
Footnotes
Acknowledgements
R.D. and X.Z. are co-first authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was partially supported by the National Library of Medicine of the National Institutes of Health under Award Numbers R01LM011829, R01LM012607, and R01LM009012, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI130460 and R01AI116794, the National Institute of Mental Health of the National Institutes of Health under Award Number P50MH113840, the Cancer Prevention Research Institute of Texas (CPRIT) Training Grant RP160015, and the National Cancer Institute under award number R35-CA197461. Also, thanks to Cerner for providing the valuable Health Facts EMR data.