
Abstract
Introduction:
Injury is the second leading cause of death worldwide, and as much as 40% of injury-related mortality is attributed to uncontrollable hemorrhage. This persists despite establishment of regionalized trauma systems and advances in the management of severely injured patients. Trauma-induced coagulopathy has been identified as the most common preventable cause of postinjury mortality.
Methods:
A review of the current literature was performed by collecting PUBMED references related to trauma-induced coagulopathy. Data were then critically analyzed and summarized based on the authors’ clinical and research perspective, as well as that reported by other institutions and researchers interested in trauma-induced coagulopathy. A particular focus was placed on those aspects of coagulopathy in which agreement among clinical and basic scientists is currently lacking; these include, pathophysiology, the role of blood components and factor therapy, and goal-directed assessment and management.
Results:
Trauma-induced coagulopathy has been recognized in approximately one-third of trauma patients. There is a vast range of severity, and the emergence of viscoelastic assays, such as thrombelastography and rotational thromboelastogram, has refined its diagnosis and management, particularly through the establishment of goal-directed massive transfusion protocols. Despite advancements in the diagnosis and management of trauma-induced coagulopathy, much remains to be understood regarding its pathophysiology. The cell-based model of hemostasis has allowed for characterization of endothelial dysfunction, impaired thrombin generation, platelet dysfunction, fibrinolysis, endogenous anticoagulants such as protein-C, and antifibrinolytic proteins. These concepts collectively compose the contemporary, but still partial, understanding of trauma-induced coagulopathy.
Conclusion:
Trauma-induced coagulopathy is a complex pathophysiological condition, of which some mechanisms have been characterized, but much remains to be understood in order to translate this knowledge into improved outcomes for the injured patient.
Introduction
Injury is the second leading cause of death worldwide, and as much as 40% of injury-related mortality is attributed to uncontrollable hemorrhage (1). In both the civilian and military setting, uncontrolled hemorrhage is the leading preventable cause of postinjury mortality (2). This persists despite establishment of regionalized trauma systems and advances in the management of severely injured patients.
Hemostasis is the physiologic cessation of bleeding achieved by the fluid and cellular phases of clotting. In the critically injured patient, often in hemorrhagic shock, coagulopathy results from a hemostatic system that is challenged by physiologic extremis. This leads to dysfunctional thrombus formation and ultimately uncontrolled bleeding. Understanding the mechanisms of hemostasis that lead to an effective control of bleeding is key in order to reduce postinjury mortality.
The purpose of this review is to examine the current knowledge gaps in trauma-induced coagulopathy (TIC) and their relevance to management.
Methods
A review of the current literature was performed by collecting PUBMED references related to TIC. Data were then critically analyzed and summarized based on the authors’ clinical and research perspective, as well as that reported by other institutions and researchers interested in TIC. A particular focus was placed on those aspects of coagulopathy in which agreement among clinical and basic scientists is currently lacking: these include pathophysiology, goal-directed assessment and management, and the role of blood components and factor therapy.
Pathophysiology
In 1954, Mario Stefanini (3) addressed the New York Academy of Medicine with a presentation reviewing the state-of-the-art knowledge on the “Basic Mechanisms of Hemostasis.”. He acknowledged how the preceding number of theories on hemostatic mechanisms had always exceeded and not always respected the confirmed experimental facts. He then admitted that the recent accumulation of new findings had almost been too rapid for their orderly incorporation into a logically working pattern, and stated that a state of “orderly ignorance” had transitioned to one of “confused enlightenment.” Stefanini concluded that the ponderous literature on hemostasis may be a classic example of the human mind’s infinite ability for abstract speculation. Our current understanding of coagulopathy may not be far from what Stefanini concluded 60 years ago.
The physiological mechanisms of hemostasis are complex, particularly in the dysregulated states of coagulopathy and inflammation in response to severe injury. Substantial knowledge has been gained by characterizing the molecules and some of the pathways that drive clot formation. However, the regulation of the clotting system and its cross talk with other adaptive mechanisms, particularly inflammation, remains poorly understood. Our attempts to manage coagulopathy have so far parted from our basic comprehension of clot formation under homeostatic conditions, expecting that extrapolating these mechanisms to simply a more severe pathologic state will explain what we observe clinically. However, the more we learn about hemostasis and the physiology of shock, it becomes evident that unique mechanisms such as certain protein functions and cell signaling exist under conditions of extremis.
The first reports of coagulopathy after injury were from studies performed at the time of the Korean and the Vietnam War, during a period when whole blood was transfused (4). These studies documented elevated prothrombin (PT) and partial thromboplastin times (PTT) in casualties, and their degree of coagulopathy correlated with the number of units of blood received. These findings were interpreted as a state of consumed factors and platelets, exacerbated by dilution derived from fluid administration and blood transfusion. In 1969, Simmons et al. (5) reported that this consumptive state was consistent with disseminated intravascular coagulation (DIC) and that treatment should be with blood component replacement. In 1982, our institution published the notion of a “bloody vicious cycle” (6), as clinical and experimental research data indicated that hypothermia and acidosis were conspicuous factors associated with early mortality in coagulopathic trauma patients (7). This “bloody vicious cycle” was subsequently referred by others as the “lethal triad” and most recently has been integrated into the concept of “iatrogenic trauma coagulopathy.” This concept was the fundamental basis of “damage control surgery” introduced in 1983 (8). Damage control surgery prioritizes early management of coagulopathy, hypothermia, and acidosis, while operating time is minimized to only control sources of significant bleeding and gastrointestinal contamination. This perspective and management prevailed for the ensuing 20 years. The components of the “bloody vicious cycle” are included in Fig. 1, which illustrates a contemporary understanding of TIC.
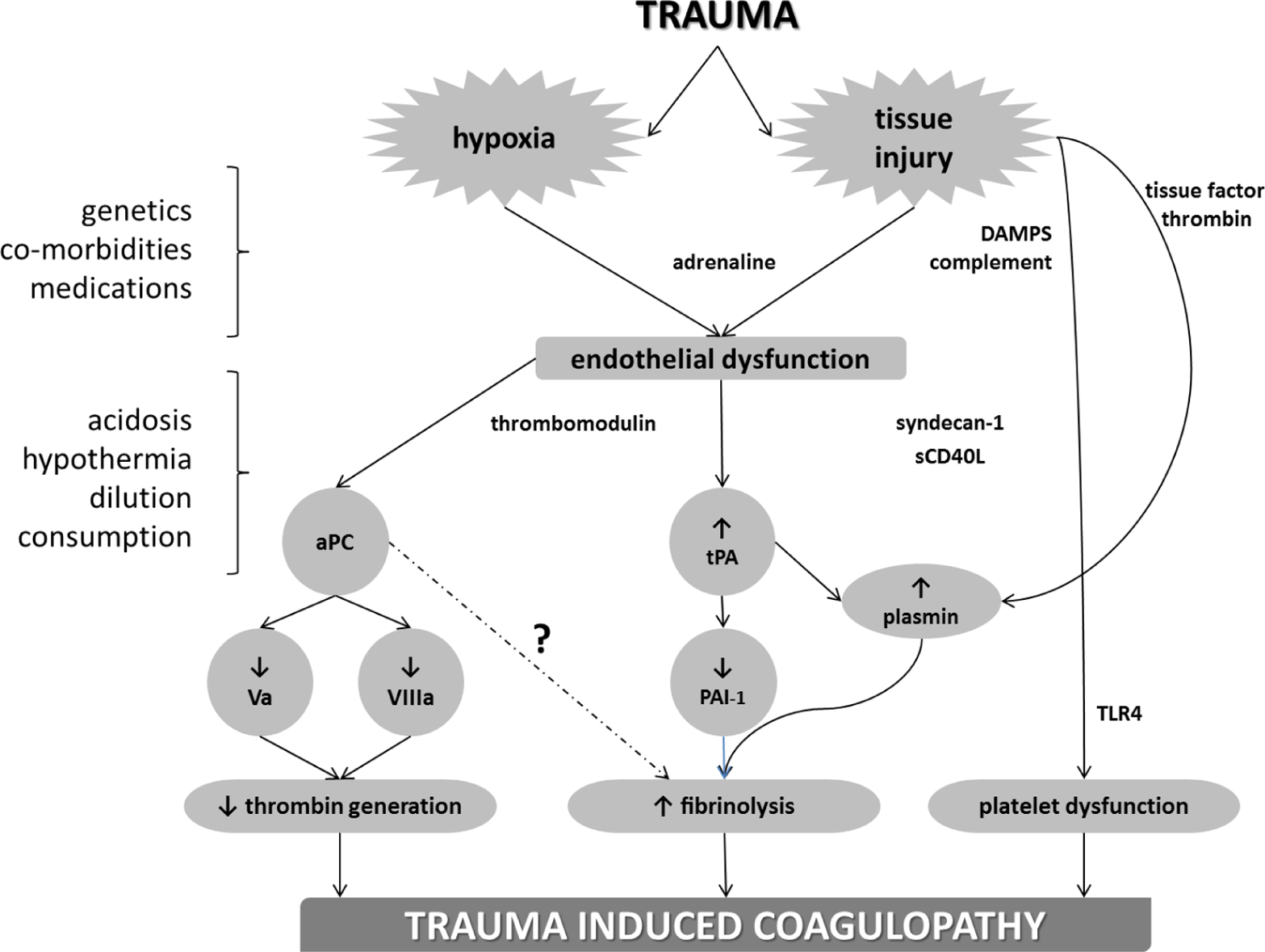
This figure represents our current understanding of trauma-induced coagulopathy. On the left-hand column are those intrinsic and acquired factors that further potentiate TIC, including acidosis and hypothermia which were initially described with coagulopathy as the “bloody vicious cycle.” Hypoxia derived from hemorrhagic shock and tissue injury appears to be synergistic in driving TIC and activation of protein-C.
Decreased concentration of circulating coagulation proteins has been quantified in healthy volunteers after administration of intravenous fluids and red blood cell transfusions (9). A correlation between the amount of resuscitative fluids and decreased circulating coagulation factor concentrations has been suggested by a clinical study (10). However, studies of this nature are subject to selection bias, given their retrospective nature. Furthermore, our group demonstrated that in an animal model of hemorrhagic shock, clotting is not compromised, as measured by viscoelastic parameters, until resuscitation with crystalloid fluids achieves 50% hemodilution in vivo (11). Although dilution with resuscitative fluids and blood transfusions exists, it is not essential for the development of TIC, and its role may only be limited to exacerbating an already compromised hemostatic system.
The current knowledge of coagulation derived from contemporary studies has progressed to a new level by abandoning the traditional teaching on hemostasis described by intrinsic and extrinsic in-vitro enzymatic cascades. Hoffman and Monroe (12) proposed a “cell-based model of hemostasis” as an in vivo model. This work describes coagulation occurring not as a “cascade,” but in three overlapping stages: initiation, amplification, and propagation of clotting, all regulated by properties of cell surfaces and their receptors. These three stages are illustrated in Fig. 2. Exposed subendothelial collagen localizes circulating platelets at the site of injury by binding to the platelet glycoprotein receptor glycoprotein-VI (GP-VI), which initially activates the platelet, however to a lesser degree than thrombin (13). Platelet activation by thrombin via the protease-activated receptors 1 and 4 (PAR-1 and PAR-4) is required for enzymatic assembly of coagulation factors on the platelet surface during the subsequent amplification and propagation phases (12). Circulating von Willebrand factor (vWF) also binds to collagen at the site of injury, and the vWF/collagen complex then adheres the platelet via the GP-Ib-V-IX receptor, further localizing platelets (13). Interestingly, the relative importance of platelet glycoproteins in the initial tethering of platelets depends on the shear rate of the vessel. At low shear rates, fibrin is the predominant ligand via the GP-IIb-IIIa receptor, whereas vWF, via GP-1b-IX-V, plays a more important role at higher shear rates (14, 15). Nevertheless, during vessel injury, the GP-VI receptor bound to collagen initially activates the platelet, causing release of adenosine-diphosphate (ADP), serotonin, and thromboxane-A2. In turn, these agonists localize and activate other platelets. Endothelial injury exposes subendothelial tissue factor expressed on fibroblasts, smooth muscle cells, and pericytes; circulating cells such as monocytes are also capable of expressing tissue factor (TF). TF then acts as a receptor and cofactor for circulating factor VII. Once bound to TF, factor VII is rapidly activated (VIIa) through mechanisms not yet completely understood but possibly involving FXa or noncoagulation proteases (12). The resulting TF/VIIa complex catalyzes activation of factor X. Thrombin is generated from the interaction between the TF/VIIa complex and Xa. This initial amount of thrombin is not sufficient to cleave fibrinogen into fibrin to produce clotting; however, it is capable of activating platelets as well as factors V and VIII during the initiation phase. In the amplification phase, activated factors V and VIII form the prothrombinase (Va/Xa) and tenase (VIIIa/IXa) complexes, respectively. Prothrombinase and tenase then enzymatically potentiate thrombin generation, into what has been described as a “thrombin burst” (13) that occurs during the propagation phase. This amount of thrombin is then capable of cleaving fibrinogen into fibrin, and maintains a positive feedback with further thrombin generation through prothrombinase and tenase. The available fibrin then forms a clot and integrates platelets by binding to the GP-IIb-IIIa receptor. In addition to platelets, red blood cells can also actively participate in thrombin generation. Whelihan and Mann (16) have described that a subfraction of red blood cells are capable of producing thrombin via the meizothrombin pathway.
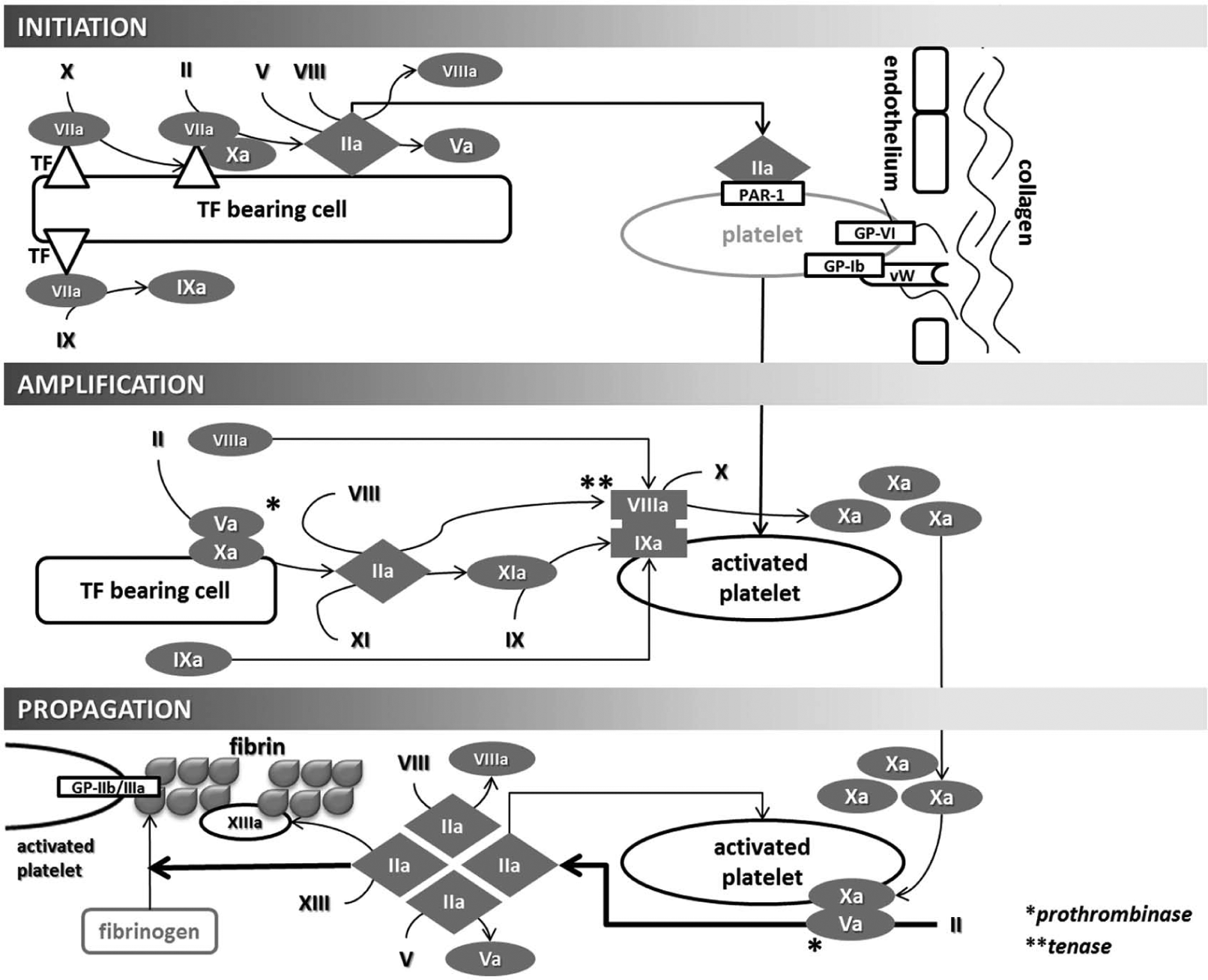
Cellular-based model of hemostasis. Initiation is triggered by TF bearing cells that activate factor VII. The TF/VIIa complex activates Xa and generates thrombin. During the initiation phase, thrombin activates factor V (which then associates with Xa to from the prothrombinase (Va/Xa) complex), factor VIII (which associates with IXa to form the tenase (VIIIa/IXa) complex during the amplification phase), and activates platelets via the protease-activated receptor (PAR-1). Simultaneously, platelets are being localized to the site of injury and activated by collagen via the platelet receptors GP Ib-IX-V (GPIb) and GP-VI. Factor IXa is also generated by TF/VIIa during the initiation phase, which can later directly participate in the propagation phase as part of the tenase complex, without requiring activation by XIa; this represents an overlap between the classically described mutually exclusive extrinsic and intrinsic pathways. During the amplification phase, the prothrombinase complex then generates thrombin that yields VIIIa and IXa, which form the tenase complex (VIIIa/IXa) on the activated platelet. The tenase complex will generate enough Xa to maintain pro-thombinase-mediated thrombin generation during the propagation phase. The amount of thrombin generated during the propagation phase is enough to cleave fibrinogen into fibrin, and also further generates Va and VIIIa (which further feeds prothrombinase and tenase), as well as XIIIa (which cross-links soluble fibrin into a stable clot).
Endogenous anticoagulant proteins have a central role in ensuring microvascular integrity during hemostasis (17, 18). Protein-C, protein-S, thrombomodulin, tissue factor pathway inhibitor (TFPI), and antithrombin (AT) have been well characterized as endogenous anticoagulants. These proteins prevent spontaneous clotting and assure that the hemostatic response is not disproportional to the amount of injury; however, physiologic extremis may lead to a pathologic unbalance. TFPI inhibits factor Xa and the VIIa/TF complex. Thrombin is directly inhibited by circulating AT, as well as by the binding of thrombin to the endothelial transmembrane glycoprotein thrombomodulin (19). Furthermore, sustained hypoperfusion is associated with increased endothelial expression of thrombomodulin as well as increased circulating soluble thrombomodulin, increasing the availability of thrombomodulin-bound thrombin (17). The thrombin/thrombomodulin complex on the surface of endothelial cells then activates protein-C (19), which is more susceptible to activation by this complex when found bound to the endothelial protein-C receptor (EPCR), as illustrated in Fig. 3. Activated protein-C (APC) modulates coagulation by cleaving peptide bonds of activated factors FVa and VIIIa, preventing their assembly into the prothrombinase (Va/Xa) and tenase (VIIIa/IXa) complexes (19). This substantially diminishes thrombin generation and its downstream effects. Thus, thrombin can be diverted from a predominantly pro-coagulant role to a pathologic anticoagulant role via excess activation of protein-C (17).
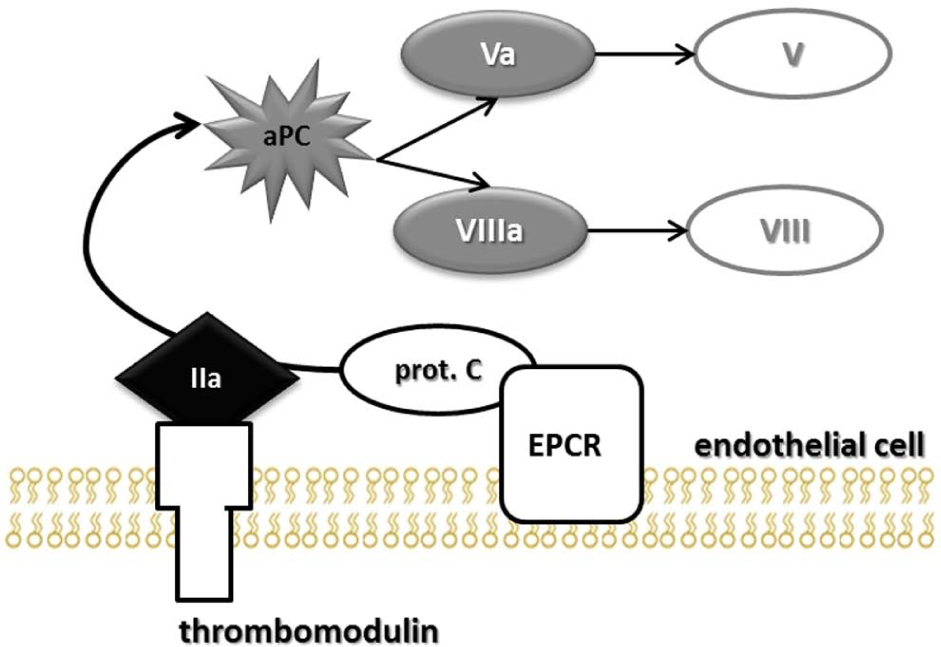
Pathway for protein-C activation. Thrombomodulin is a transmembrane endothelial protein that binds thrombin. The thrombomodulin/thrombin complex is then responsible for protein-C activation (aPC). The endothelial protein-C receptor (EPCR) binds circulating protein-C to the endothelial surface, making it more susceptible to activation by the thrombomodulin/thrombin complex. aPC cleaves activated factors Va and VIIIa into their inactive forms V and VIII.
The characterization of endogenous anticoagulants has brought further insight into the pathophysiology of TIC, parting from the long-standing notion that coagulopathic bleeding observed after injury was consistent with DIC. DIC is a clinico-pathological syndrome characterized by systemic generation of fibrin, leading to organ failure due to microvascular occlusion, and concomitant consumption of coagulation factors and platelets leading to bleeding (20). Gando et al. propose that DIC yields both a fibrinolytic, or hemorrhagic, and an antifibrinolytic, or thrombotic, phenotype which sometimes overlap (23). The diagnosis is based on a scoring system consisting of the presence of a decreased platelet count, prolonged PT, decreased fibrinogen, and elevated D-dimer level, in the setting of clinical suspicion (21). Although it is understandable that most severely injured trauma patients will meet some of these diagnostic criteria, contemporary studies have demonstrated that coagulopathic bleeding can exist in the absence of factor consumption. Brohi et al. (17) documented an elevated PT in trauma patients upon emergency department (ED) arrival; however, there was no evidence of exhaustion of thrombin, given that levels of prothrombin fragments were not elevated. Furthermore, the pathophysiology of DIC is based on the presence or absence of fibrinolysis (22, 23), yet not all coagulopathic trauma patients have demonstrable fibrinolysis, nor do they all have hypofibrinogenemia (24). These findings argue against consumption as an indispensable underlying mechanism for TIC. It must be recognized that DIC may complicate the course of trauma patients at any point in their management; however, its development is usually in the later setting of overwhelming sepsis, rather than initially after injury and/or hemorrhagic shock (23). This suggests that DIC and TIC are distinct pathophysiological entities with similar clinical variables that often overlap.
TIC: Activated Protein-C
With this background of the basic science proposal of hemostasis, the clinical translation has emerged in a step-wise fashion, as shown in Fig. 1. The work of Brohi et al. (17) in 2007 hypothesized that an acute traumatic coagulopathy, observed within 1 h of injury, was caused by tissue hypoperfusion resulting in activation of the anticoagulant protein-C pathway, because patients without tissue hypoperfusion as measured by base deficit (BD) were not coagulopathic, and prolongation of PT and PTT was only observed with an increased BD. This relationship was attributed to increased protein-C activity measured upon ED arrival. These same investigators prevented prolongation of PTT in a mouse model of trauma and hemorrhagic shock by antibody-mediated inhibition of protein-C (25). Furthermore, during this same model, synergism of trauma and hemorrhagic shock were required for prolongation of PTT, as tissue trauma or hemorrhagic shock was unable to prolong PTT individually (25). In a study that used a statistical analysis technique of pattern finding and data reduction, known as principal components analysis, patterns of TIC based on coagulation factor and endogenous anticoagulant levels were identified. Two distinct patterns of TIC were described: (1) global coagulation factor depletion, which was associated with penetrating injury as well as injury severity, and predicted coagulopathy and mortality, and (2) increased protein-C activation and fibrinolysis, which were associated with hemorrhagic shock, and predicted multiple organ failure, acute lung injury, pneumonia, and mortality. These data suggest distinct and possibly overlapping patterns of TIC; however, their biological mechanisms remain to be understood, particularly regarding fibrinolysis.
TIC: Hyperfibrinolysis
Clinical studies have identified that in those coagulopathic patients with the highest hemorrhage-related mortality, fibrinolysis is a conspicuous factor (24, 26). Fibrinolysis using conventional laboratory assays, such as euglobulin lysis time, was suspected to be present in 15%−20% of trauma patients over a broad range of injury severity (27). The advent of viscoelastic assays has greatly expanded our understanding of the role of fibrinolysis in TIC. The more severe forms of fibrinolysis (>15% of clot lysis 30 min after researching maximum clot strength) can predict mortality, in which fulminant lysis (complete clot degradation within 30 min) is associated with 100% mortality (24). Recent studies identifying postinjury fibrinolysis using thrombelastography (TEG) have refined the degree of clot lysis that is associated with adverse outcomes from >15% to >3% (28, 29). However, by measuring plasmin-anti-plasmin levels (30) in trauma patients, it has been argued that TEG misses fibrinolytic patients. Whether this degree of fibrinolysis detected by antiplasmin (AP) levels and not by TEG is of any clinical consequence has yet to be established. The key unresolved issue is what represents physiological, or protective, versus pathologic, or coagulopathic, fibrinolysis in the injured patient.
While we continue to refine our understanding of fibrinolysis in trauma, it is important to understand the mechanisms keeping the fibrinolytic system in check. Plasminogen is the central pro-enzyme of fibrinolysis, which is depicted in Fig. 4. Plasminogen interacts with numerous cellular receptors that have functions beyond fibrinolysis, but its primary role when activated into plasmin by tissue plasminogen activator (tPA) or urokinase is to breakdown cross-linked fibrin (31, 32). The lysine binding domains (LBD) of plasminogen have specificity for the lysine on carboxy-terminal region of cross-linked fibrin (31), localizing plasminogen to the forming clot. Regulation of plasminogen is possible by multiple proteins related to this interaction. For example, tPA also contains LBDs that localizes it to plasminogen bound to fibrin, activating plasminogen into plasmin and catalyzing fibrinolysis (33). Plasminogen/plasmin’s major inhibitor, antiplasmin (AP), also utilizes the LBD. This inhibitor can directly bind to this region and prevent plasminogen interaction with fibrin in addition to forming a nonreversible inhibition when bound to plasmin (34). AP can also be incorporated into cross-linked fibrin by factor XIIIa reducing the amount of susceptible lysine regions to be cleaved by plasmin (34). Thrombin activatable fibrinolysis inhibitor (TAFI) is a carboxy-peptidase that cleaves the lysine residues of partially plasmin-digested fibrin, reducing plasmin binding and activity (35). It is not a surprise that pharmaceuticals such as tranexamic-acid (TXA) and aminocaproic-acid that interfere with LBDs can reduce fibrinolysis (36). Regulation of plasminogen activation is another point of control. Plasminogen activator inhibitor-1(PAI-1) is a serine protease inhibitor (SERPIN-E1) that functions as the principal inhibitor of (tPA) (37). Hyperfibrinolysis has not yet been fully integrated into the currently proposed mechanisms of TIC, as a mechanistic link is lacking. In-vitro studies have adjudicated antifibrinolytic properties to protein-C (38), and low PAI-1 levels have been reported in trauma patients with increased protein-C activity (17). This appears to be an association, but no mechanism of the interaction between these two proteins has been described. A potential explanation is based on observations of elevated tPA/PAI-1 complexes during endothelial ischemia (37). Excessive tPA derived from endothelial ischemia during injury may bind all available PAI-1; this tPA/PAI-1 complex is subsequently cleared by the liver, favoring un-inhibited tPA and promoting unregulated fibrinolysis. The important discoveries in fibrinolysis will come from differentiating physiologic lysis driven by hemostasis during tissue injury to maintain vascular patency, from pathologic and dysregulated fibrinolysis resulting in uncontrolled hemorrhage.
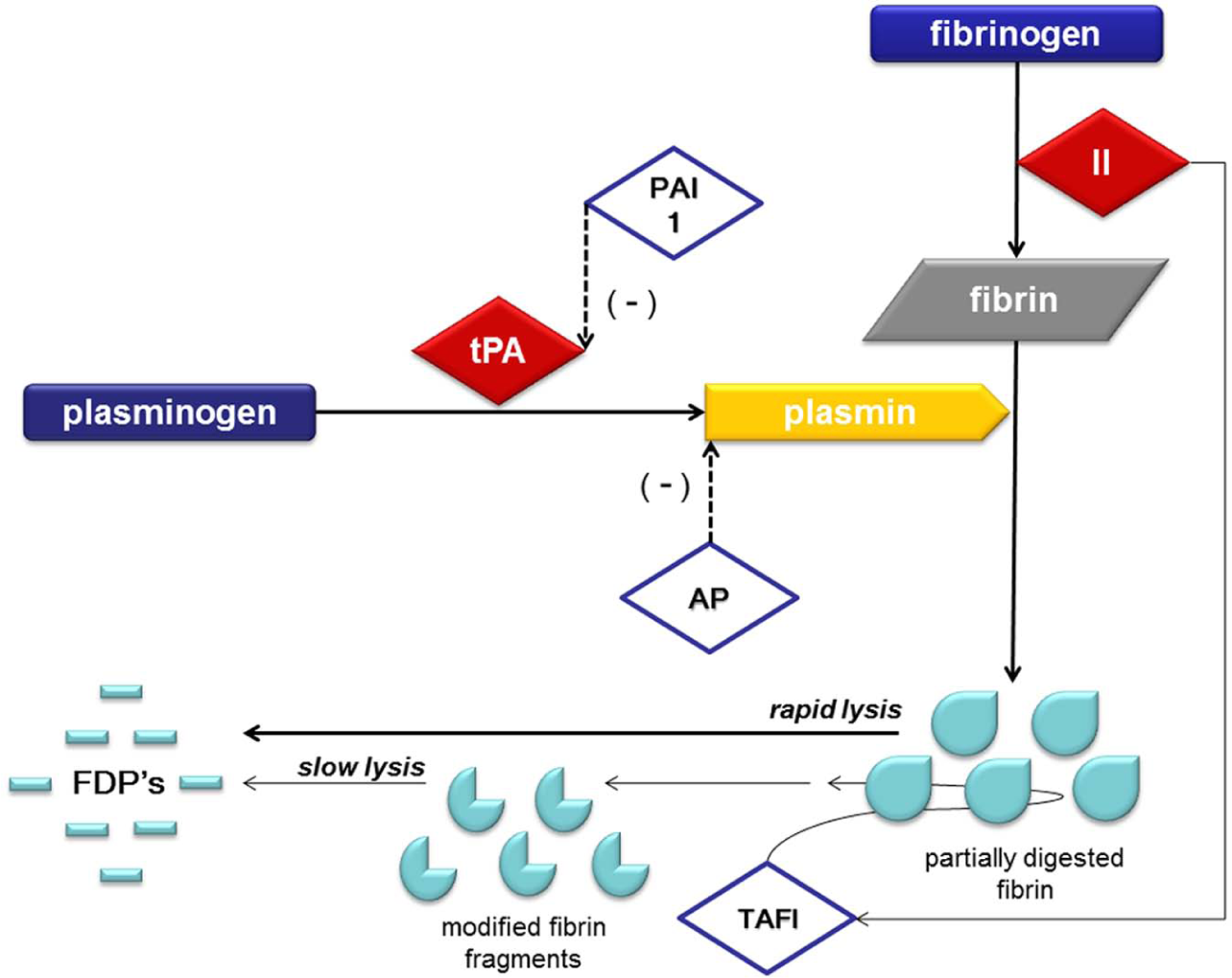
Mediators and inhibitors of fibrinolysis. In circulation, plasminogen adopts a closed, activation-resistant conformation. Upon binding to fibrin, plasminogen adopts an open form that can be converted into active plasmin by tissue plasminogen activator (tPA). Plasmin achieves fibrinolysis by cleaving fibrin into fibrin dimer products (FDPs). FDPs are quantified by the D-dimer laboratory assay. tPA activity is inhibited by plasminogen activator inhibitor 1 (PAI-1) and most circulating tPA is bound to this inactivator. Thrombin activatable fibrinolysis inhibitor (TAFI) down regulates fibrinolysis by modifying fibrin with removal of C-terminal lysines, making fibrin less susceptible to plasmin cleavage and decreasing the rate of fibrinolysis. Antiplasmin (AP) forms a stoichiometric complex with soluble plasmin directly inhibiting its activity.
TIC: Platelet Dysfunction
Early platelet dysfunction may also play a role in TIC. Kutcher et al. (39) demonstrated that upon ED arrival, 45% of trauma patients had compromised platelet function by multiplate impedance aggregometry. This early platelet dysfunction has also been described in experimental animal models of trauma/hemorrhagic shock and traumatic brain injury (TBI) (40), where it was present as early as 15 min after injury. Our group recently quantified the degree of platelet-agonist receptor inhibition in trauma patients (41), utilizing TEG-based platelet mapping. The viscoelastic strength of the clot for each platelet agonist is measured while inhibiting the fibrin contribution to the clot. We found 86% inhibition of ADP mediated platelet aggregation in trauma patients compared to 4% in healthy volunteers, and 44% arachidonic acid (AA) inhibition compared to 0.5% in healthy volunteers. To explain this platelet dysfunction, Johansson et al. (42) proposed a concept of platelet activation with subsequent exhaustion, occurring early after injury. This is based primarily on their observation of elevated CD40L, a soluble platelet ligand, in severely injured patients. Platelet dysfunction has also been associated with TBI by several studies (39, 40). In a study from our trauma patient population, ADP receptor inhibition correlated strongly with severity of TBI. Furthermore, the percentage of ADP inhibition distinguished between survivors and nonsurvivors of TBI. It is also unclear if platelet dysfunction after injury and shock is independent of the dysfunction of circulating coagulation proteins previously described.
TIC: Endothelium
The endothelium is also an active participant in the pathophysiology of TIC. This has recently been evidenced by the work of Johansson et al. (43), which measured plasma levels of syndecan-1, an endothelial glycocalyx protein, in trauma patients upon ED arrival. In this study, an elevated level of soluble syndecan-1 was associated with a prolonged PTT, increased protein-C activity, and circulating catecholamines, and was an independent predictor of mortality in trauma patients. Elevated adrenaline levels were independently associated with increased PTT and syndecan-1. These investigators propose that shock and the accompanying sympatho-adrenal activation and hypoperfusion may directly contribute to glycocalyx degradation from the endothelium. These effects of circulating catecholamines on the endothelium present a challenge for experimental animal models of TIC, which are performed under anesthesia, limiting this sympatho-adrenal activation seen in trauma patients. Much remains to be known regarding the role of the endothelium in the development of coagulopathy, with the main challenge being the lack of assays that can directly evaluate dynamic endothelial activity and its interaction with the fluid phase of coagulation.
Coagulation Assessment
Optimal management of coagulopathy starts with prompt diagnosis. Time is the main catalyst of coagulopathy, as the bloody vicious cycle perpetuates itself with every minute untreated. Clinical suspicion based on known risk factors for coagulopathy such as estimated injury severity score (ISS) >15, BD >6, and systolic blood pressure (SBP) <90 mmHg upon presentation (44) should prompt an immediate work-up and goal-directed treatment if needed. We initiate our massive transfusion protocol (MTP) based on field and ED vital sign criteria established by the Research Outcomes Consortium (ROC), with the addition of high risk injury patterns such as penetrating torso wounds, unstable pelvic fractures, or abdominal ultrasound with evidence of bleeding in more than one region. Our institution’s MTP is described in Fig. 5.
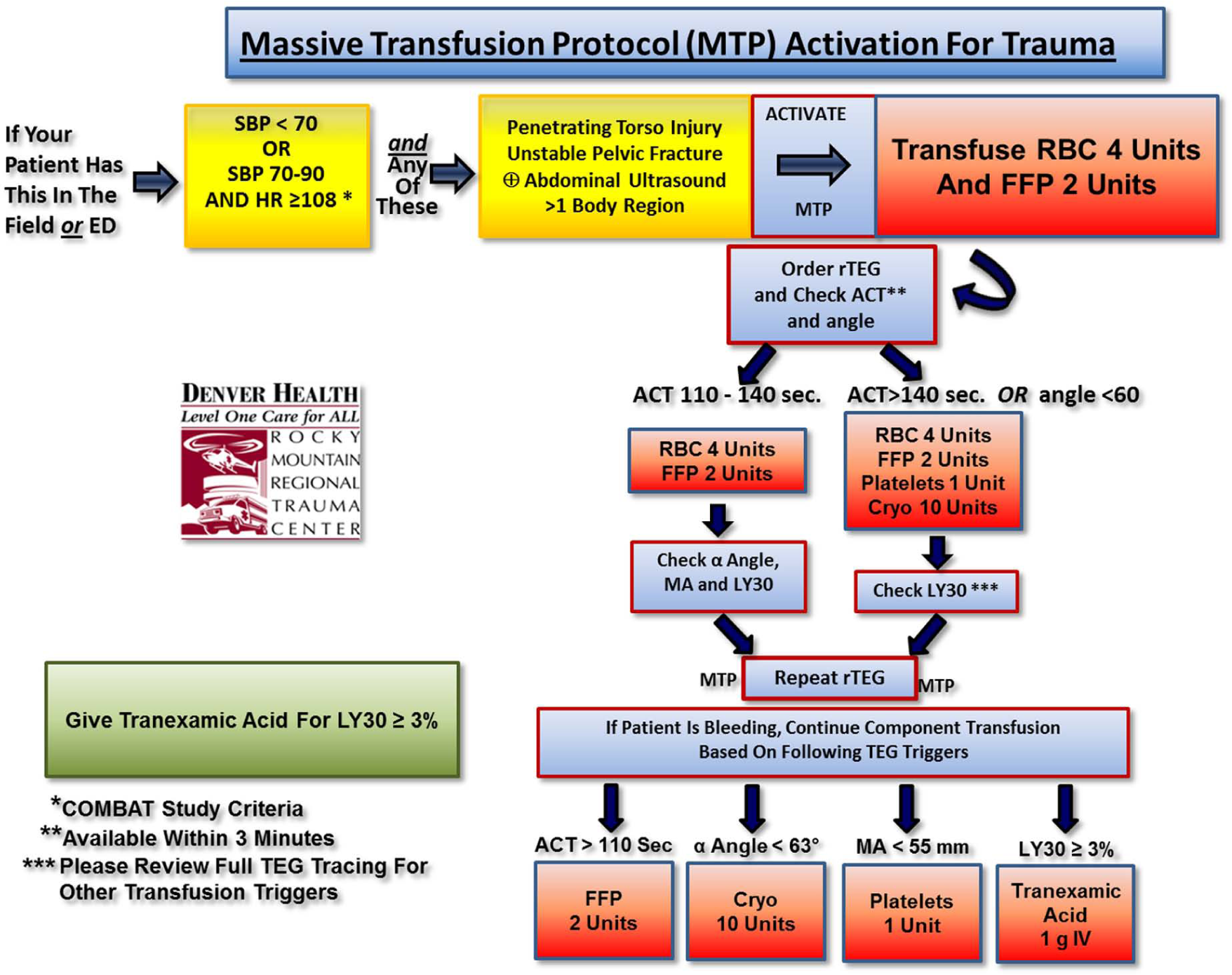
Algorithm of massive transfusion protocol incorporating goal-directed therapy.
Optimally, a coagulation assessment should be performed on the field, and evolving viscoelastic technologies will soon allow for field point-of-care assessment. Until a decade ago, the diagnosis of TIC was achieved with traditional coagulation tests such as PT, international normalized ratio (INR), and PTT. These tests are still considered the standard of care in many institutions for diagnosing TIC. However, it is important to recognize that PT/INR and PTT were initially developed for the screening of heritable coagulopathies such as hemophilia, and subsequently used to monitor anticoagulant therapy (45). The end point for these tests is the time, in seconds, until the earliest formation of fibrin is detected by optical or electro-mechanical means. These assays do not assess the evolution of the clot beyond the formation of the first strands of fibrin. Furthermore, they have shown to correlate poorly with bleeding risk in elective general and vascular surgeries (46). Fibrinogen and D-dimer levels are surrogate markers of clotting factor consumption and fibrinolysis respectively; however, these are also nonspecific in assessing hemostasis of the injured patient (45). Furthermore, the gold-standard assay of fibrinolysis, euglobulin lysis time, requires at least 4 h for results to become available.
The earliest studies to describe coagulopathy after injury used PT and PTT to establish this diagnosis. Several studies have used the following thresholds to define coagulopathy: PT >18 s (47), INR >1.5 (48), PTT >60 s (47), or any of these values at a threshold of 1.5 times their reference value (49). In trauma patients, the prevalence of a prolonged PT is higher, but prolongation of the PTT is more specific for diagnosing TIC. MacLeod et al. (50) reported a prolonged PT and PTT in 28% and 8% of trauma patients, respectively, with an adjusted odds ratio for mortality of 1.35 for PT and 4.26 for PTT prolongation. In another study, a more liberal INR cutoff (>1.2) increased its sensitivity to identify patients with greater transfusion requirements and increased mortality. However, these patients had a greater ISS compared to other studies, suggesting that this lower INR value may be a more appropriate cutoff for patients with more severe injury (ISS > 15). In fact, PT and PTT may be more biomarkers of injury severity rather than mediators of TIC.
A platelet count along with a hemoglobin/hematocrit should be obtained on all trauma patients upon ED arrival. A decreased platelet count in trauma patients with active bleeding has been traditionally considered critical at less than 100,000/µL, as it has been associated with poor outcomes and mortality after trauma (51, 52), as well as with poor neurological outcomes in patients with TBI (52). However, a platelet count provides no information on platelet function. Using serial functional analyses with aggregometry, conserved platelet function and activation has been demonstrated in survivors following trauma, while nonsurvivors had significant dysfunction (53). Platelet aggregometry remains the gold standard for assessment of platelet function; however, its results are not immediately available, and it has not been clinically evaluated in trauma resuscitation.
It is unclear whether TIC can occur in patients with a normal PT/INR and PTT. Anecdotal reports exist of trauma patients with significant bleeding secondary to fibrinolysis, and a normal PT/INR and PTT. Thus, clinical suspicion is critical for prompt identification and management of coagulopathy, in spite of having traditional coagulation tests within normal range. In addition to the criteria defined for activation of our MTP, several scoring systems have been developed in order to identify those patients that will develop TIC. These include the Trauma-Associated Severe Hemorrhage (TASH) score, Assessment of Blood Consumption (ABC) score, and the McLaughlin score. Nunez et al. (54) compared these scoring systems and found significantly higher TASH (13 versus 6), McLaughlin (3.4 versus 2.4), and ABC (2 versus 1) scores in patients who required massive transfusion compared with patients who did not. However, no significant predictive differences were identified between these scores. Interestingly, none of these scoring systems include coagulation parameters, and fundamentally represent the same concept of the bloody vicious cycle our group described in 1981 (7).
As classic coagulation tests have shortcomings when used to diagnose and monitor coagulopathy in the trauma patient, viscoelastic assays such as TEG and ROTEM have emerged in the last decade as a novel application of a well-established technology. Hellmut Hartert conceived TEG in Germany, at the Heidelberg University School of Medicine in 1948. During the 1950s, TEG was applied increasingly throughout Europe and validated as a reliable assessment of hereditable coagulopathies, anticoagulant therapy, presence of thrombocytopenia, and fibrinolysis (55). The availability of TEG in the United States was limited until the 1980s, when its clinical utility was confirmed in liver transplantation for diagnosis of fibrinolysis during the anhepatic phase (56). TEG was subsequently used during cardiopulmonary bypass, and in the last decade, to identify and monitor TIC (57). This assay has gained widespread acceptance as it was demonstrated to allow differentiation of coagulopathy from surgical bleeding.
The TEG analyzer and the tracing it yields are illustrated in Fig. 6A. The methodology of this technology has been well described in the literature (57). In brief, TEG provides data on the viscoelastic properties of the clot in a dynamic way, from the earliest mechanical resistance provided by the first strands of fibrin formed to loss of strength secondary to lysis (57). The parameters that quantify the evolution of these stages of clot formation are described in Table 1. Of note, traditional clotting assays such as PT/INR and PTT fail to quantify clotting beyond the initiation phase, which corresponds to the R-time or TEG-activated clotting time (TEG-ACT) (58). Blood coagulation in TEG is initiated by exposure of whole blood to the foreign surface of the TEG cup. Addition of coagulation activators such as kaolin or TF will expedite generation of results. Most TEG parameters can be obtained within 10 min when using TF as an activator, allowing for prompt decision making in the critically injured patient. This assay is referred to as rapid-TEG and is the standard TEG obtained in the trauma bay. In the ROTEM device, another viscoelastic assay based on similar principles as TEG, graphical output appears similar to that obtained with TEG. Characteristics and variables of ROTEM are described in Fig. 6B and Table 1 (59).
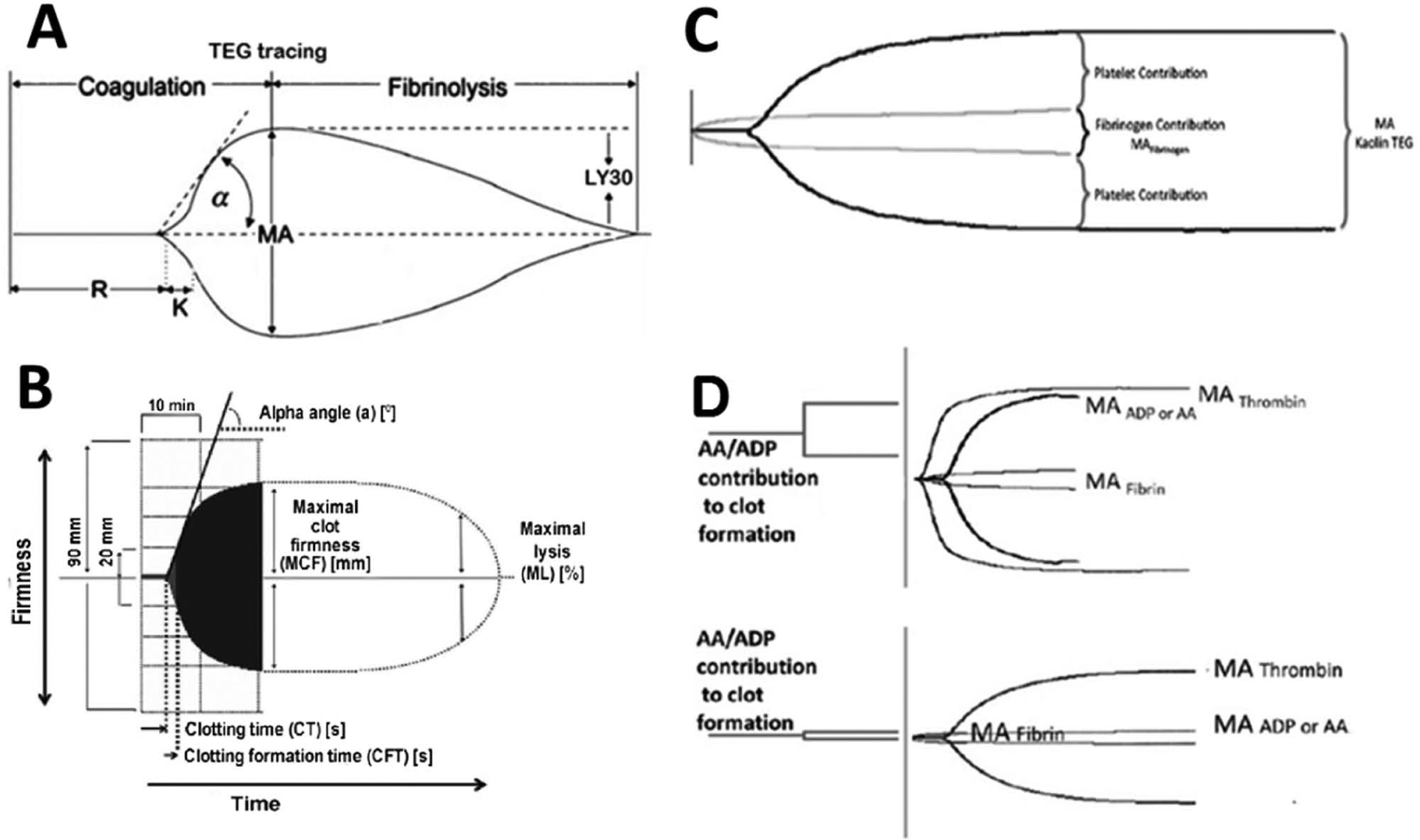
Classic illustration of the (A) thrombelastogram and (B) ROTEM viscoelastic tracings. Parameters for these tracings are described in Table 1. (C) Functional fibrinogen tracing demonstrating fibrin and platelet contribution to clot strength. (D) TEG platelet mapping. The amplitude (mm), depicted on the y-axis, in response to platelet agonists maximum amplitude-adenosine-diphosphate (MA-ADP) or maximum amplitude-arachidonic acid (MA-AA) is compared with the maximum hemostatic activity (maximum amplitude (MA)-Thrombin) minus fibrin contribution (MA-Fibrin). The superior tracing represents a normal contribution of platelet’s to the clot, and the inferior tracing represents compromised platelet function.
Viscoelastic parameters of the stages of clot formation.
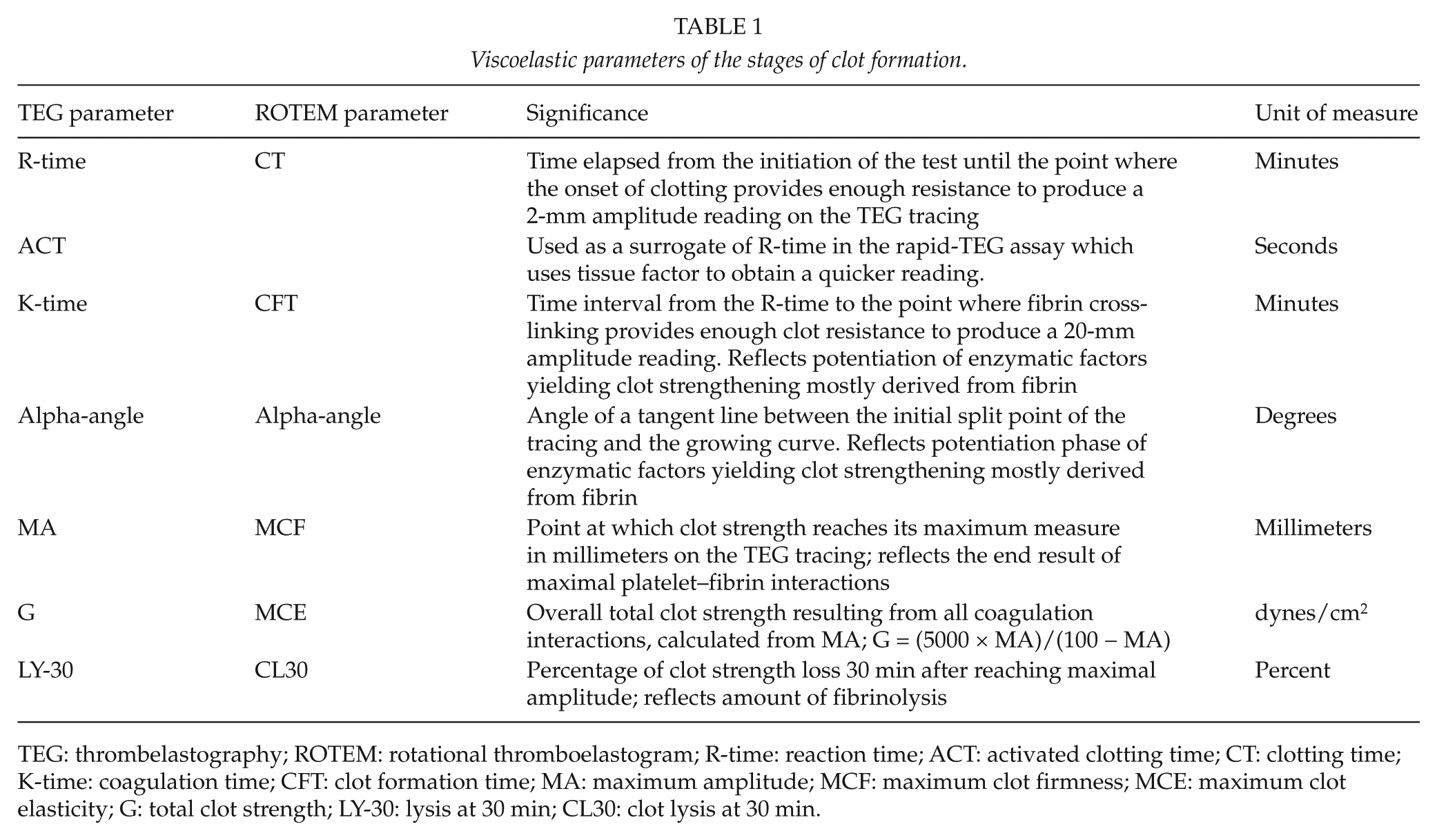
TEG: thrombelastography; ROTEM: rotational thromboelastogram; R-time: reaction time; ACT: activated clotting time; CT: clotting time; K-time: coagulation time; CFT: clot formation time; MA: maximum amplitude; MCF: maximum clot firmness; MCE: maximum clot elasticity; G: total clot strength; LY-30: lysis at 30 min; CL30: clot lysis at 30 min.
The thrombelastogram has been validated against other coagulation assays that are specific to each phase of the coagulation process. By generating velocity curves of clot formation derived from TEG parameters, investigators have demonstrated a correlation between these parameters and thrombin generation as measured by thrombin-antithrombin (TAT) levels (60). The TEG parameters, derived from a single measurement of whole blood coagulation, are not independent parameters but rather a continuum of blood coagulation with interactions between all components. For instance, thrombin liberates fibrinopeptides from fibrinogen, allowing their association with other soluble fibrin molecules, and subsequently thrombin-activated factor XIII converts soluble into cross-linked fibrin (13). This impacts the rate of clot strengthening as represented by the alpha-angle and K-time. Furthermore, thrombin exerts known effects on platelet function via PAR-1, and interactions with factors VIII and von Willebrand; accordingly, such changes mainly impact maximum amplitude (MA) and shear elastic modulus strength (G). Regarding fibrinolysis, data from our institution demonstrated that severe fibrinolysis, defined by a TEG LY-30 (lysis at 30 min) >15%, is associated with massive transfusion requirements, coagulopathy, and hemorrhage-related death (26). However, others have suggested that plasmin-antiplasmin levels are a more sensitive marker of fibrin breakdown (30). This supports a lower TEG LY-30 threshold of >3% as an indication for treatment, more recently recommended by the Houston group (28), as well as ours (29).
As a point-of-care instrument, the availability of real-time TEG and ROTEM tracings in the ED, operating room (OR), or intensive care unit (ICU), as they are generated, allows for prompt pattern recognition, that provides the clinician with bedside information for critical decision making.
The TEG assay has also been modified in order to further enhance its ability to isolate and quantify the contribution of available fibrinogen to hemostasis. Although the TEG parameters alpha-angle and K-time correlate with fibrinogen concentration levels, they do so poorly with the more sensitive Clauss assay of fibrinogen (61). A functional fibrinogen TEG (FF-TEG) assay has been developed as a more sensitive method to quantify fibrinogen function, as depicted in Fig. 6B. Briefly, the FF-TEG isolates the fibrinogen contribution to clot strength by using a monoclonal glycoprotein IIb-IIIa receptor antagonist to eliminate platelet contribution to clotting. Our group has demonstrated that FF-TEG correlates strongly with the Clauss assay, and allows for differentiation of the contribution of both platelets and fibrinogen to the clot’s strength (62).
Clot formation via the standard TEG and ROTEM assays is dependent on thrombin, mostly because clotting initiation is via exposure of whole blood to the foreign surface of the cup, and because of the addition of activators such as kaolin or TF. Given that thrombin is the most potent platelet activator (13), dysfunctional platelets due to ischemia during trauma/hemorrhagic shock or even under the influence of the platelet inhibitors acetyl-salicylic acid and clopidogrel may still generate enough clot strength to yield an MA within normal range due to the presence of thrombin. In order to better identify platelet dysfunction with TEG, a platelet mapping assay, PM-TEG, has been designed, which relies on the measurement of clot strength by MA to enable a quantitative analysis of platelet function. PM-TEG uses heparin as an anticoagulant to eliminate thrombin activity in the sample. Reptilase and factor XIIIa are used to generate a cross-linked fibrin clot and eliminate the fibrin contribution to the clot strength. The contribution of the P2Y or thromboxane platelet receptors to clot formation can be measured by the addition of either ADP or AA, respectively, as platelet agonists, as illustrated in Fig. 6C. The use of PM-TEG to guide goal-directed resuscitation remains to be validated clinically.
Parallel to the TEG, ROTEM has also been validated in diagnosing TIC. This technology has emerged mostly in Europe. Although the sensitivity of their parameters to detect coagulopathy is comparable between the two (63), the numeric output of each of the parameters are not interchangeable, despite having similar interpretations. In a comparison study, it was noted that TEG and ROTEM values are not interchangeable (64). European and American investigators studied a total of 184 trauma patients from three different countries prospectively by performing paired TEG and ROTEM analyses. Limits of agreement between most TEG and ROTEM parameters exceeded the preset clinically acceptable deviation of 10% (64). This study concluded that interchangeability between TEG and ROTEM values is limited, and that development and validation of separate treatment algorithms for the two devices are required.
It is important to highlight the limitations of viscoelastic point-of-care assays such as TEG and ROTEM. First, these assays do not reflect most interactions occurring between the fluid phase of coagulation and the endothelial cell surface; this dynamic is clearly present in the coagulopathic patient; however, it remains to be understood. Second, platelet inhibition/dysfunction may not be evident with the standard assays, unless thrombin is inhibited and platelet agonists are utilized in TEG-PM and ROTEM assays. Third, TEG and ROTEM results are operator dependent and subject to sampling and/or processing errors, as well as intersampling variability.
Hemostasis Management
Given that hemorrhage-related mortality after injury occurs primarily within the first 2 h after injury, it is imperative that management of coagulopathy begins at the moment of clinical suspicion. As a general principle, trauma patients should be initially approached with the universal A-B-C assessment and management strategy, regardless of their coagulation status. This allows for airway control, oxygenation, hemodynamic resuscitation, and control of bleeding to take precedence, as efforts to achieve hemostasis will be futile if these principles are not addressed in a timely fashion. Particular attention to treatment of hypothermia while resuscitation is ongoing is also imperative.
In the coagulopathic patient, hemostatic resuscitation needs to be achieved concomitantly with hemodynamic resuscitation. Red blood cell transfusion not only improves perfusion and oxygen carrying capacity but also provides sufficient cellular phospholipid surfaces to sustain the enzymatic assembly responsible for thrombin generation (16). As a general consensus, it is recommended that a hemoglobin level above 10 g/dL should be maintained in injured patients with active bleeding. Recently, there has been a shift toward reduced administration of crystalloid volume and use of blood products to achieve hemostasis and oxygen delivery while benefiting from the volume also provided by them. Kutcher et al. (65) reported that systematization of such practices under an institutional MTP was associated with decreased blood product use, and when adjusting for age and injury characteristics, with a 5.6% reduction in mortality. MTPs are either based on fixed plasma:red blood cell (RBC), cryoprecipitate:RBC, and/or platelet (PLT):RBC ratios agreed upon institutions, or goal directed by viscoelastic point-of-care assays. Tapia et al. (66) reported a survival benefit with a goal-directed TEG-based MTP compared to a fixed 1:1:1 (RBC:fresh frozen plasma (FFP):PLT) MTP in penetrating trauma patients requiring >10 units of blood. It has been suggested that the benefit reported from MTPs is not related to delivering certain ratios of RBCs and plasma or platelets, but rather by breaking down the logistic barriers between the blood bank and the patient, enabling the bedside clinician. MTPs allow for immediate processing of blood products by the blood bank and their systematic delivery to the patient’s bedside. They create uniform treatment strategies to be easily followed by all medical providers across all disciplines. However, caution must be exercised, as liberal criteria for activation of these protocols, or lack of monitoring of the progression of these patients’ hemodynamic and hemostatic status, may lead to indiscriminate administration of blood products. MTPs continue to vary widely between trauma centers and ratios of plasma:RBC range from 1:1 to 1:10 (48, 67). With low plasma ratios, treatment of coagulopathy becomes delayed, and ultimately a greater volume of blood is required. However, it remains unclear at what point does administration of higher plasma ratios is no longer beneficial. The recognition of this problem has led to a widespread re-evaluation of transfusion protocols and the approach to damage control resuscitation is now institution dependent.
No prospective randomized trials are yet available to provide a standardized approach to transfusion for the trauma population. The ratio of blood products at which this benefit is optimal remains a matter of debate. Based on recent data, what is certain is that coagulopathic trauma patients meeting criteria for massive transfusion benefit from early plasma and platelet administration, while being judicious with crystalloid administration (65). This benefit appears to be independent of the ratio of blood product used, as all studies that report improved outcomes with a particular ratio gave plasma and/or platelets earlier (68). Furthermore, a similar 24-h survival benefit has been observed in patients who receive either increased plasma:RBC or PLT:RBC ratios (69), making this survival benefit not product specific. Searching for an ideal ratio is subject to variation in logistics of blood banking and provider delivery as well as survival bias. It is important to underscore this survival bias to which all of these studies are subject to. Their retrospective nature allows for patients who have died early during resuscitation to be excluded; patients may have had longer survival not because they received more plasma, but rather received more plasma because they survived long enough to do so (70). Parenthetically, in 1982, our group proposed that when transfusing the trauma patient in hemorrhagic shock, the first unit of plasma should be administered in the ED concomitantly with the first four units of RBCs, based on a survival benefit observed at the time (6). Management of TIC at our institution is based on principles of damage control resuscitation and goal-directed therapy, as described in Fig. 5.
Goal-directed resuscitation should allow for immediate re-assessment of the patient’s coagulation in a timely fashion that allows critical decision making at the bedside. The point-of-care availability of viscoelastic assays, such as TEG and ROTEM, allows for such a goal-directed approach. These goal-directed resuscitation protocols will likely emerge as the new standard, but only single-center studies are currently available (71, 72).
Based on positive experience reported from retrospective studies on use of whole blood (WB) for resuscitation in the US military (73), a recent clinical trial was designed by Cotton et al. (74) to address whether WB was superior to blood product component therapy. In this civilian, single-center study, WB was not superior to blood component therapy in reducing transfusion volumes or decreasing 24-h and 30-day mortality in severely injured patients predicted to receive massive transfusion. A benefit was seen only in a sensitivity analysis where patients with brain injury were excluded, where the use of WB resulted in significantly reduced transfusion volumes compared to blood component therapy. Due to institutional regulations, this study used modified WB by leukoreduction, which also clears platelets, and stored these WB units at 1°C to 6°C, which further decreases platelet function. Because of this, patients receiving this modified WB received supplementation with an apheresis platelet unit for every 6 units of modified WB. This is in contrast to the published military studies, which used fresh, warm WB. These authors report this might be responsible for the discrepancy in results between the positive retrospective military experience and this clinical trial.
In addition to repletion of coagulation factors by transfusion, several pharmaceutical hemostatic agents are available for the treatment of severe coagulopathy in the injured patient. These products include fibrinogen concentrate (FC), recombinant factor VIIa, prothrombin complex concentrate (PCC), desmopressin, and antifibrinolytic agents such as TXA, aminocaproic-acid, and aprotinin.
Fibrinogen levels are decreased early after injury in coagulopathic trauma patients. Thus, supplementation of fibrinogen has been advocated in the management of TIC, as provided by plasma and cryoprecipitate transfusion. Although plasma does contain fibrinogen, its low concentration requires a large amount of volume for transfusion; hence, cryoprecipitate has remained the main source of fibrinogen supplementation at most institutions. Recently, European experience has suggested that the use of FC is effective in the management of TIC, as evidenced by improved fibrinogen function based on the ROTEM fibrinogen thromboelastometry (FIBTEM) assay (75). FC label use in Europe allows for its use both in congenital and acquired coagulopathies; however, in the United States, its use is restricted to congenital coagulopathies.
PCC is a factor concentrate of factors II, VII, IX, and X, originally developed for hemorrhagic complications of hemophilia. The clinical use of PCC has been well studied in the reversal of warfarin anticoagulation, since PCC is preferentially rich in the vitamin K-dependent clotting factors. Some centers have used PCC to correct TIC, and preliminary studies in animal models of hemorrhagic shock are promising (76). In a retrospective review, the use of PCC therapy in trauma patients lead to a significant correction in INR and concomitant reduction in blood product transfusion, however with no improvement in mortality. PCCs are potent pro-coagulants, and as such, the possibility of associated thromboembolic complications should be carefully considered. The benefit of PCC in trauma patients has not yet been studied prospectively, particularly comparing its effectiveness to that of plasma and/or cryoprecipitate transfusion.
Transfusion of blood products, particularly plasma, has been linked to development of subsequent inflammatory conditions such as transfusion-related acute lung injury. In this setting, the use of specific factor component replacement agents, such as FC or PCC, to supplement plasma and/or cryoprecipitate transfusion in an attempt to reduce transfusion volumes, has been proposed. Factor replacement with FC and PCC should not be considered equivalent to plasma or cryoprecipitate transfusion, as they do not contain certain proteins present in these blood products, particularly factors V, VIII, and XIII. Furthermore, plasma transfusion may confer additional benefits than FC and PCC beyond factor replacement (77). Plasma is a third-generation resuscitation fluid. Like first-generation crystalloids, plasma is iso-osmolar with blood and contains all of the cations and anions present in blood. Like the second-generation colloid resuscitation fluids based on albumin alone, or nonhuman polysaccharides such as large dextrans and starches, it has high oncotic pressure (28 mmHg versus 3 mmHg in 0.9% saline). In addition to coagulation factors, it also contains albumin, transferrin, immunoglobulins, apolipoproteins, and protease inhibitors. A proteomic analysis of human plasma has revealed several potentially cytoprotective proteins to be highly concentrated, including protease inhibitors (78). Perhaps most compelling is emerging evidence for the role of endothelial glycocalyx degradation in the pathogenesis of coagulopathy (43), as previously described in this review. Endothelial glycocalyx disruption has also been associated with release of danger signals such as histone-complexed DNA and high-mobility group box-1 (43). Additionally, the endothelial glycocalyx can also regulate neutrophil adhesion (79). Recent experimental work has shown that resuscitation with plasma, compared to crystalloid, attenuates endothelial glycocalyx disruption following hemorrhagic shock (80). Neither FC nor PCC has been validated prospectively against cryoprecipitate or plasma in the management of TIC. Limiting the management of TIC with FC or PCC, and not using plasma transfusion, may eliminate the potential benefit provided by these still not well-characterized benefits of plasma beyond restoration of hemostasis.
Recombinant factor VIIa was initially developed for the treatment of hemophilia and other congenital factor deficiencies (81). The recognition of exposed TF binding to activated factor VII as the principal trigger of clot formation after trauma led to interest in using recombinant factor VIIa for the management of TIC. Recombinant human factor VIIa is an adjunctive treatment for TIC but should be reserved for salvage therapy, as no clinical trial has validated its effectiveness in trauma. Its use is mostly limited to promptly reverse coagulopathy of therapeutically anticoagulated patients with severe hemorrhage or TBI. When used, it is important to correct acidosis, hypothermia, thrombocytopenia, and hypofibrinogenemia, in order for it to be effective.
An interest in antifibrinolytic therapy has been renewed because of the recent recognition of hyperfibrinolysis following severe trauma. In the Clinical Randomization of an Antifibrinolytic in Significant Hemorrhage (CRASH-2) trial, an absolute mortality reduction of 1.5% was identified in patients receiving empiric TXA compared with placebo (82). No coagulation assays were used to describe the degree of coagulopathy and/or fibrinolysis of the patients enrolled, nor to characterize the effect of such drug. Enrollment criteria included adult trauma patients within 8 h of injury with significant hemorrhage (SBP < 90 mmHg or heart rate (HR) > 110 beats/min), or those who were considered to be at risk of significant hemorrhage. The characteristics of the population studied are the primary criticism of this trial; only 50% of patients met inclusion criteria and only half of those received a blood transfusion. Furthermore, there was no significant reduction in RBC transfusion requirements in the treatment arm of the study. The subsequent Military Application of Tranexamic-acid in Trauma Emergency Resuscitation (MATTERs) study evaluated outcomes in 896 patients who were treated with TXA or not (83). Unadjusted mortality rates were significantly reduced for treated compared to nontreated casualties, 17% versus 24% respectively. Other antifibrinolytic agents include aminocaproic-acid and aprotinin, but these have not been prospectively evaluated in patients with TIC. In a comprehensive analysis of the data of all clinical studies using TXA as an antifibrinolytic in trauma patients performed by Napolitano et al. (84), only a modest effect on the overall population treated was observed; all-cause mortality was reduced from 16.0% to 14.5% (number-needed-to-treat, 67), and the risk of death caused by bleeding overall was reduced from 5.7% to 4.9% (number-needed-to-treat, 121). TXA’s greatest impact on mortality was in those in the severe shock group (SBP < 75 mmHg). Furthermore, the mechanism by which TXA reduced mortality in the CRASH-2 trial remains unclear given that fibrinolysis and coagulation assessments were not part of the study design. This calls for caution of indiscriminate use of antifibrinolytic drugs and raises the question of whether coagulation assessments should be performed prior to their administration.
Finally, desmopressin, a drug developed for the treatment of von Willebrand’s disease (85) and uremic bleeding, has shown to improve platelet function. There is insufficient clinical evidence to support the use of desmopressin in the trauma population except in those patients with preexisting bleeding diatheses (86). Preliminary animal studies show that desmopressin improves hypothermia and acidosis-induced platelet dysfunction, but clinical validation is lacking (87).
Footnotes
Declaration of Conflicting Interests
None.
Funding
This research was funded by National Institutes of Health; NIH-P50 #GM49222, NIH-T32 #GM008315.