
Abstract
Background and Aims
CpG islands of the promoter region of some genes are methylated in pancreatic cancer tissue and the detection of this methylation has been suggested to be useful in the diagnosis of various cancers. The aim of this study was to investigate whether the detection of methylated CpG islands in plasma can be used in the diagnosis of pancreatic cancer.
Material and Methods
Plasma DNA was collected from patients with pancreatic cancer, chronic pancreatitis, and healthy controls. The methylation status of six genes, UCHL1, NPTX2, SARP2, ppENK, p16, and RASSF1A, was checked by methylation-specific PCR and was subsequently confirmed by direct sequencing after bisulfite treatment.
Results
CpG island methylation was detectable in 13 of 16 patients (81.3%) with pancreatic cancer, 1 of 29 healthy controls (3.5%), and 8 of 13 patients with chronic pancreatitis (61.5%). The mean number of genes with CpG island methylation was 1.6 ± 1.2 in pancreatic cancer, 0.04 ± 0.19 in healthy controls, and 1.2 ± 1.1 in chronic pancreatitis. Among six genes, p16 was more specifically methylated in pancreatic cancer compared with chronic pancreatitis (p = 0.016). The methylation status was not correlated with smoking history, tumor size, or cancer stage.
Conclusions
The detection of methylated genes in the plasma may have a role in differentiating between pancreatic cancers and healthy controls but not between pancreatic cancer and chronic pancreatitis.
Keywords
INTRODUCTION
Pancreatic cancer is one of the most aggressive cancers and, accordingly, has the lowest survival rate of any solid cancer throughout the world. Despite recent advances in diagnosis and treatment, patients with this devastating disease have a very poor prognosis. Despite tremendous efforts to improve the overall prognosis of patients with pancreatic cancer, infiltrating ductal carcinoma of the pancreas remains one of the most deadly human cancers. As a result, mortality from this disease almost parallels its incidence, with a 5-year survival rate of < 10% (1). More than 80% of pancreatic cancers are too far advanced and non-resectable because they are rarely diagnosed at an early stage in affected patients.
Because of difficulties in early diagnosis and highly aggressive malignant behavior, only 10–20% of pancreatic cancers can be surgically resected with curative intent at the time of diagnosis, and most patients experience local recurrence and metastasis even after surgical resection. Although gemcitabine can prolong survival of patients and is the current standard therapy for advanced pancreatic cancer, < 5% of patients survive 5 years after the initial diagnosis and the median survival duration is about 6 months due to a high degree of inherent and acquired chemoresistance (2).
Usually, percutaneous fine-needle aspiration (FNA) cytology or biopsy and endoscopic retrograde cholangiopancreatography (ERCP)-guided pancreatic duct brush cytology are used for pathologic confirmation. The reliability of percutaneous FNA cytology or biopsy is approximately 80% (range, 65–95%) (3). However, percutaneous FNA and ERCP-guided pancreatic duct brush cytology are invasive procedures, and sometimes it is anatomically difficult to approach the mass of the pancreas because of surrounding major vessels. In addition, there is a risk of seeding cancer cells during the procedure. Thus, percutaneous FNA has usually not been required in patients with resectable pancreatic cancer. Although ERCP-guided pancreatic duct brush cytology is less invasive and presents no risk of seeding, the reliability of cytologic analysis is not high enough (4). Therefore, methods detecting surgically resectable tumors could significantly reduce deaths from this aggressive disease.
During pancreatic cancer tumorigenesis, many genetic and epigenetic alterations occur. Among the oncogenes, K-ras mutations occur in 90% and AKT2 mutations in 10–20%. Among tumor suppressor genes, INK4a/p16 is inactivated in 80–95%, p53 in 75%, TGF-β/DPC4 in 50%, and BRCA2 in 5–10% (5).
One of epigenetic alterations is DNA methylation, which involves promoter CpG island methylation and thereby silencing involved genes (6). In pancreatic cancer, CpG islands of p16 are methylated in 43% (7), preproenkephalin (ppENK) in 90% (8). and Ras association domain family protein 1 (RASSF1A) in 64% (7). Recently, Sato et al. (9) reported that ubiquitin carboxyl-terminal esterase L1 (UCHL1), neuronal pentraxin II (NPTX2), and secreted apoptosis related protein 2 (SARP2) are methylated in 100%, 98%, and 95% of primary pancreatic cancer tissues, respectively. In contrast, NPTX2, SARP2, CLDN5, reprimo, LHX1, TJP2, CDH3, and ST14 are completely unmethylated in normal pancreatic ductal epithelia (9). Previous studies have reported that tumor DNA is released into the circulation and is enriched 4–40 fold in plasma and serum of cancer patients compared to healthy controls (10). Based on these observations, studies have shown that it is possible to detect tumor-specific DNA in the plasma of pancreatic cancers patients. In pancreatic cancers, K-ras mutations could serve as a good marker to detect tumor DNA in the plasma (11).
Chronic pancreatitis is a risk factor for pancreatic cancer. Despite the recent advances in diagnostic methods, the differential diagnosis of pancreatic cancer from chronic pancreatitis is occasionally difficult. If we could detect the methylation of plasma DNA secreted from pancreatic cancer, it could be used in the early diagnosis of pancreatic cancer and in the differential diagnosis of pancreatic cancer from chronic pancreatitis.
The aim of this study was to investigate whether the detection of CpG island methylation in the plasma by methylation-specific polymerase chain reaction (PCR) could be used in the diagnosis of pancreatic cancer. We investigated the methylation frequency of candidate genes in the plasma of patients with pancreatic cancer and the diagnostic potential of detecting methylated genes in the plasma of patients with pancreatic cancer.
MATERIALS AND METHODS
PATIENTS AND SAMPLE COLLECTION
Twenty-nine patients with unresectable, locally advanced or metastatic pancreatic cancer or chronic pancreatitis were enrolled between January and September 2004 at the Seoul National University Hospital. Informed consent was obtained from all patients and this study was carried out in accordance with the Helsinki Declaration. Pancreatic cancer was confirmed by pathologic examination of tissues obtained by percutaneous FNA or biopsy or surgery. The diagnosis of chronic pancreatitis was based on a typical history of recurrent abdominal pain plus calcification in the pancreas or ductal lesions typical to chronic pancreatitis. Healthy controls were defined as subjects without evidence of chronic pancreatitis or other cancers. Sixteen patients with pancreatic cancer were enrolled (nine males, seven females and the mean age of 63.9 ± 9.8 years) and 13 patients with chronic pancreatitis (12 males, one female and the mean age of 49.5 ± 11.8 years). Samples from twenty-nine healthy controls (11 males, 18 females and the mean age of 40.7 ± 12.0 years) were also included for the analysis.
CELL CULTURE AND DNA PREPARATION
For the methylation analysis of six genes in a panel of pancreatic cancer cell lines, four human pancreatic cancer cell lines, T-24, A549, Capan-2, and MiaPaCa-2, were obtained from the Korea Cell Line Bank (Seoul, Korea) and grown in DMEM or RPMI (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum. Cell lines were incubated under standard culture conditions. The cells were suspended in 200 μL of buffer containing trypsin-EDTA. The cells were vortexed and centrifuged at 800 rpm for 5 minutes. The nuclear pellet was resuspended in 200 μL of PBS. The supernatant was quantified using the QiAamp® DNA Blood Mini Kit (Qiagen, Seoul, Korea).
SAMPLE COLLECTION AND PLASMA DNA EXTRACTION
Ten mL of plasma samples were collected from patients with pancreatic cancer, chronic pancreatitis, and healthy controls, centrifuged at 1,800 rpm, and plasma samples were collected in EDTA-containing tubes. Genomic DNA was extracted from plasma using the QiAamp® DNA Blood Mini Kit according to the manufacturer's recommendations.
METHYLATION-SPECIFIC POLYMERASE CHAIN REACTION (MSP)
The DNA methylation patterns in the CpG islands of the target genes were determined by detecting chemical conversion of unmethylated cytosine to uracil with sodium bisulfite treatment and subsequent PCR amplification (12). Ninety μL of eluted DNA samples were treated with sodium bisulfite using an EZ DNA Methylation Kit™ (Zymo Research, Orange, CA, USA) for 16 hours at 50 °C. Three μL of bisulfite-treated DNA was amplified using primers specific for either methylated or unmethylated DNA. Primers were designed to detect the sequence differences between methylated and unmethylated DNA as a result of bisulfite modification and each primer pair contained at least four CpG sites to provide optimal specificity.
PCRs were carried out in a reaction volume of 25 μL using components of the GeneAmp® PCR Gold Buffer (Perkin Elmer, Foster City, CA, USA). Each PCR reaction mixture consisted of 0.4 mmol of each primer; 1 unit of Taq polymerase (AmpliTaq Gold®; Perkin Elmer); 200 μmol dNTP; 1.5 mmol MgCl2; and GeneAmp® 10 × PCR Gold Buffer. Three μL of treated plasma DNA solution (200 ng) was used in each MSP reaction. Amplifications were carried out in a GeneAmp® PCR System 2700 (Perkin Elmer Cetus, Norwalk, CT, USA). Thermal cycling was initiated with a first denaturation step of 95°C for 5 minutes. The thermal profile for the PCR was 95°C for 30 seconds, 55–62°C for 40 seconds for annealing, 72°C for 40 seconds for extension, and a final extension of 7 minutes at 72°C. Data obtained during 40–45 cycles of amplification were analyzed. All of the assays were performed at least twice.
To identify methylation in pancreatic cancer, we studied six candidate genes (UCHL1, NPTX2, SARP2, ppENK, p16, and RASSF1A) that had been reported to be heavily methylated in their promoter region; ubiquitin carboxyl-terminal esterase L1 (UCHL1), neuronal pentraxin II (NPTX2), secreted apoptosisrelated protein 2 (SARP2), ppENK, p16, and RASSF1A.
We first performed MSP on these six genes in four pancreatic cancer cell lines (Capan-2, MiaPaCa-2, T-24, and A549) to compare the results with the corresponding MSP data of plasma DNA.
DIRECT SEQUENCING OF THE AMPLIFIED PCR PRODUCTS
PCR products were purified using a Zymoclean Gel DNA Discovery Kit™ (Zymo Research). Purified PCR products were quantified by agarose gel electrophoresis and a UV spectrophotometer. Purified PCR products were sequenced using a BigDye® Sequencing Kit (v1.1; Applied Biosystems).
Direct sequencing was performed as follows: The cycle sequencing reaction was accomplished by the addition of template DNA (11 μL), primer (1 μL [3.2 pmol]), and BigDye® terminator reaction mixture (8 μL). Thermal cycling was performed on a GeneAmp 9600 thermal cycler (PE Applied Biosystems) with the following protocol: 96°C for 10 seconds, 50°C for 5 seconds, and 60°C for 4 minutes, for a total of 35 cycles. Subsequently, unincorporated dye terminators were removed by ethanol precipitation. The resulting products were suspended in 4 μL of loading dye (5 volumes of deionized formamide plus 1 volume of 25 mM EDTA [pH 8.0] with blue dextran [50 mg/mL]). The reactions were electrophoresed on an ABI 377 automatic sequencer (Global Medical Instrumentation, Inc., Ramsey, Minnesota, USA). Sequences were analyzed using sequence navigator software (Applied Biosystems) and compared with MSP results.
STATISTICAL ANALYSIS
Statistical significance of positive frequencies for each gene with CpG island methylation in pancreatic cancer, chronic pancreatitis, and healthy control was analyzed using the Pearson chi-square test to assess the diagnostic usefulness detecting DNA methyation in the plasma. The Mann-Whitney U test was performed to analyze differences in the CpG island methylation frequencies for each gene among pancreatic cancer, chronic pancreatitis, and healthy control. All experiments were performed more than three times. Data are expressed as the means ± SE. Statistical analysis was performed using SPSS software (SPSS 12.0K for Windows; SPSS, Seoul, Korea). A p < 0.05 was considered statistically significant.
RESULTS
CLINICAL CHARACTERISTICS OF PATIENTS
The cancer stages of pancreatic cancer patients were stage I in one patient, stage III in eight patients, and stage IV in seven patients. The mean tumor size was 3.7 ± 1.4 cm. Four pancreatic cancer patients had a smoking history > 10 pack years.
The underlying etiology of chronic pancreatitis was alcohol in eight patients and idiopathic in five patients. Nine patients with chronic pancreatitis had parenchymal calcifications in the pancreas.
METHYLATION ANALYSIS OF SIX GENES IN A PANEL OF FOUR PANCREATIC CANCER CELL LINES
UCHL1, NPTX2, SARP2, ppENK, p16, and RASSF1A were confirmed to be methylated in at least one of the pancreatic cell lines by using MSP analysis. Methylation of UCHL1, NPTX2, and SARP2 was detected in 50% of CpG islands of Capan-2 and MiaPaCa-2 pancreatic cancer cell lines, ppENK in 25% of MiaPaCa-2, and p16 and RASSF1A in 25% of T-24 and A549.
MSP ANALYSIS IN THE PLASMA
MSP analysis for plasma gene methylation in pancreatic cancer and chronic pancreatitis revealed methylation in varying frequencies (Fig. 1).
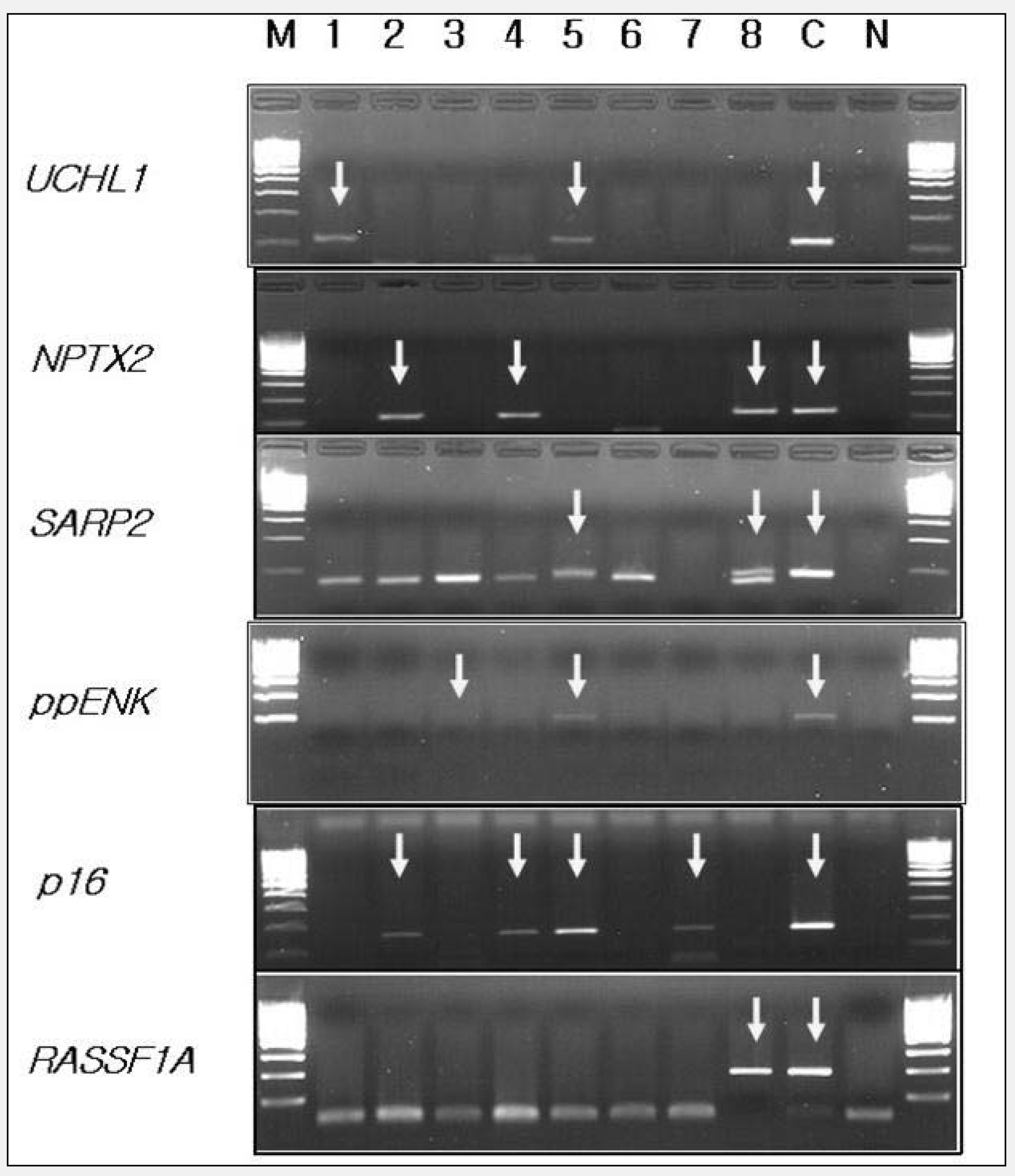
Methylation-specific PCR analysis for gene methylation. Lane 1, 2, 3, 6 were the plasma samples from the chronic pancreatitis and lane 4, 5, 7, 8 from the pancreatic cancer patients. Lane M was 100 bp DNA marker, lane C was positive control cell line. The PCR products in the lanes marked by arrows indicated the presence of methylated templates of each gene.
The results of DNA genomic sequence analyses coincided with MSP results. The frequency of methylated CpG islands from total CpG islands in each methylated gene was appeared variously (Fig. 2).

DNA sequence analysis for the detection of CpG islands methylation. The methylated PCR products were sequenced after bisulfite treatment to confirm their methylation status. The CG in the square box indicate methylated CpG island. UCHL1, NPTX2, SARP2, ppENK, p16, and RASSF1A genes showed methylation in varing frequencies.
DNA methylation in the plasma of patients with pancreatic cancer were detected in 4 of 16 patients (25.0%) for UCHL1 gene, 6 of 16 (37.5%) for NPTX2, 5 of 16 (31.3%) for SARP2, 4 of 16 (25.0%) for ppENK, 5 of 16 (31.3%) for p16, and 1 of 16 (6.3%) for RASSF1A. Methylation of in the plasma of patients with chronic pancreatitis were detected in 2 of 13 (15.4%) for UCHL1, 4 of 13 (30.8%) for NPTX2, 3 of 13 (23.1%) for SARP2, 2 of 13 (15.4%) for ppENK, 2 of 13 (15.4%) for p16, and 1 of 13 (7.7%) for RASSF1A. Methylation in the plasma of healthy controls was detected in 1 of 29 (3.5%) for p16 only. The percentage of methylated CpG islands in each gene ranged from 21.4% to 100% in patients with pancreatic cancer and chronic pancreatitis (Table 1).
Percentage of methylated CpG islands of the promoter region in each gene in plasma samples in patients with pancreatic cancer (PC) and chronic pancreatitis (CP)
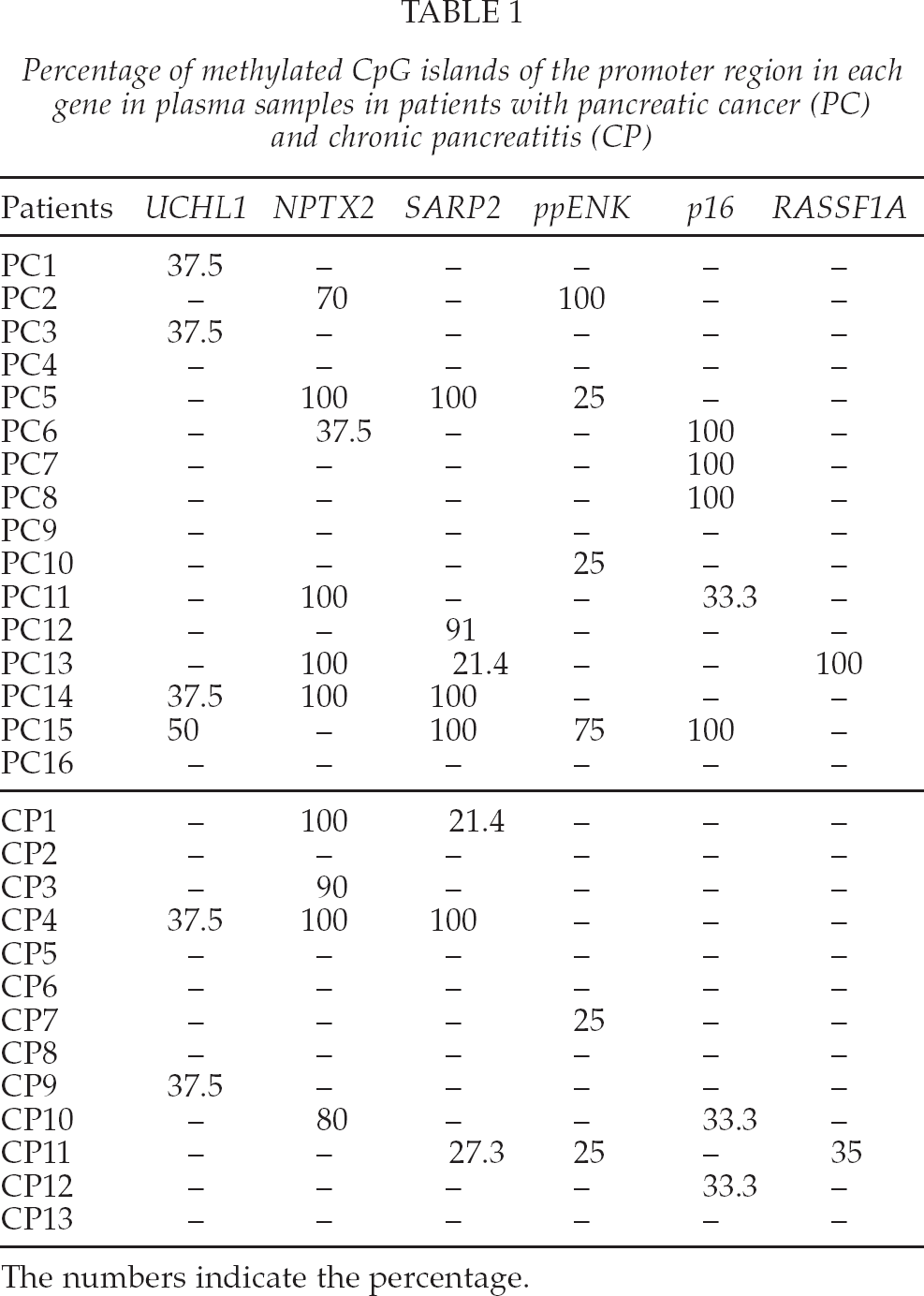
The numbers indicate the percentage.
The detection of methylation in at least 1 of 6 genes was possible in 13 of 16 patients with pancreatic cancer (81.3%), 8 of 13 chronic pancreatitis (61.5%), and in 1 of 29 healthy controls (3.5%). The detection rate was significantly higher in the pancreatic cancer compared with normal controls (p < 0.05), but the difference was not statistically significant between pancreatic cancer and chronic pancreatitis.
The average number of methylated genes among six genes in each group was 1.6 ± 1.2 in pancreatic cancer, 1.2 ± 1.1 in chronic pancreatitis, and 0.04 ± 0.19 in healthy controls. The average number of methylated genes was significantly higher in the pancreatic cancer compared with normal controls (p < 0.05), but the difference was not statistically significant between pancreatic cancer and chronic pancreatitis.
A heavy methylation (> 50% methylation) was more frequent in patients with pancreatic cancer compared with chronic pancreatitis (Fig. 3).
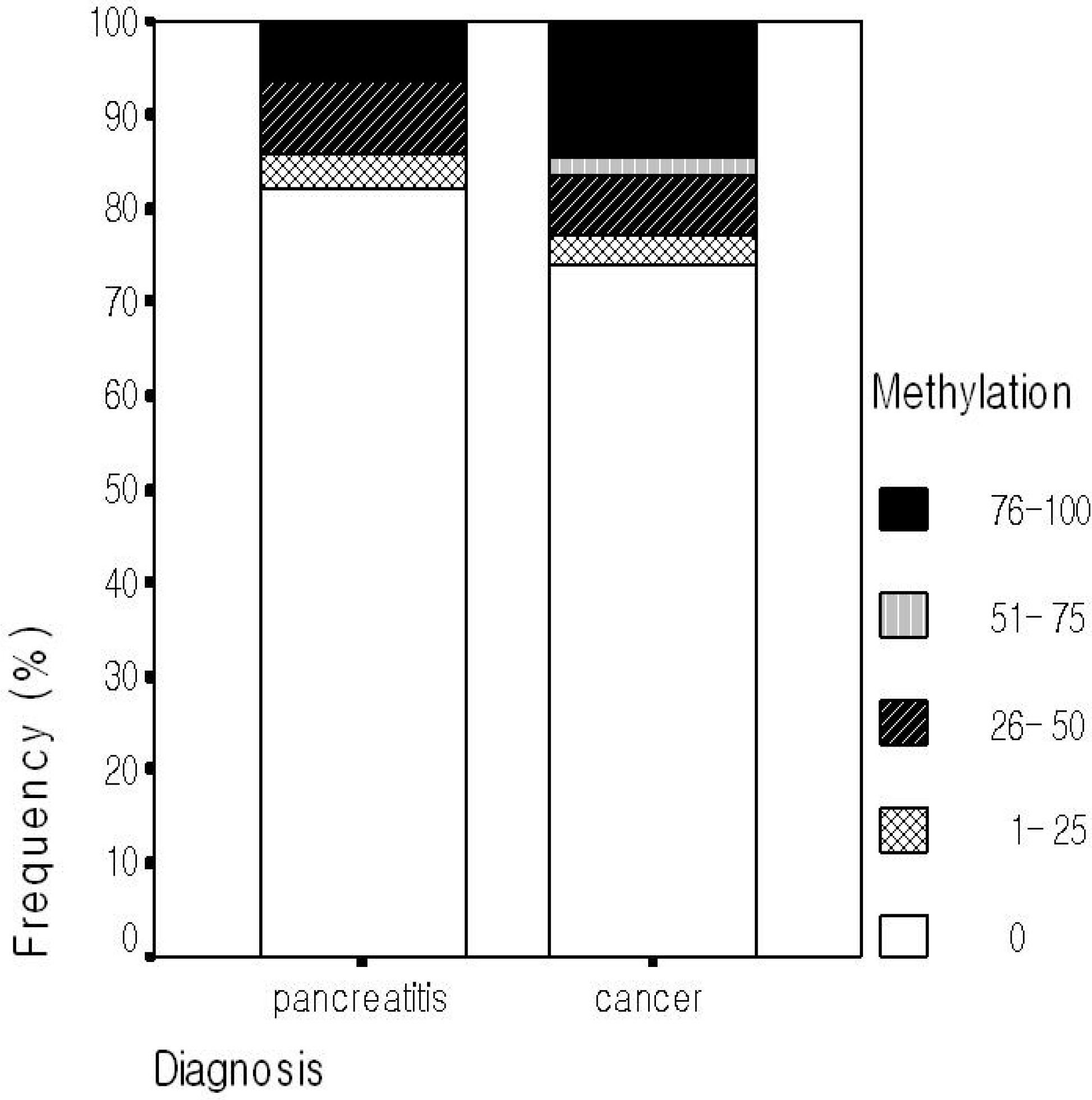
Comparison of methylation status of all CpG islands tested in the plasma between pancreatic cancer and chronic pancreatitis. Heavy methylation (more than 50% methylation) was more frequent in pancreatic cancer than chronic pancreatitis (p = 0.125).
CLINICAL CORRELATION WITH DNA METHYLATION
Methylation status of the genes in the plasma were analyzed according to cancer stage and tumor size. Methylation in at least one CpG island was detected in any stages. Detection rate was highest in stages IV. However, the percentage of methylation was not different according to tumor size. Methylation status was not affected by the history of a smoking in pancreatic cancer patients.
METHYLATED GENE AS A POSSIBLE TUMOR MARKER
The rate of methylation in pancreatic cancer tends to be higher in p16 compared with chronic pancreatitis (p = 0.075; Table 2). The proportion of methylated CpG islands in p16 was significantly higher in pancreatic cancer than in chronic pancreatitis (p = 0.016; Table 3).
Comparison of proportion of CpG islands methylation in each gene between pancreatic cancer and chronic pancreatitis
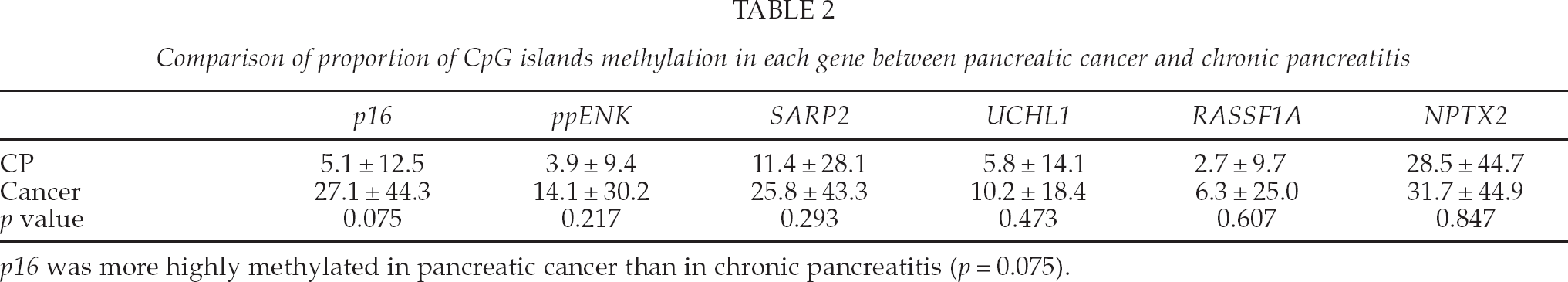
p16 was more highly methylated in pancreatic cancer than in chronic pancreatitis (p = 0.075).
Comparison of proportion of methylated CpG islands among total CpG islands in the promoter region of each gene between pancreatic cancer and chronic pancreatitis
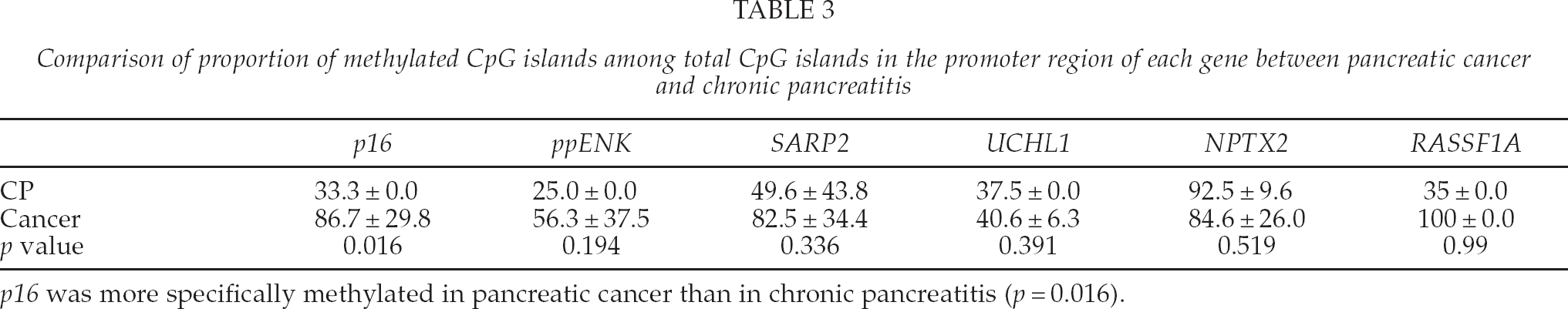
p16 was more specifically methylated in pancreatic cancer than in chronic pancreatitis (p = 0.016).
DISCUSSION
Although this is the preliminary study using small number of patients, the results suggest that the pattern of gene methylation in the plasma was rather different between the patients with pancreatic cancer and healthy controls. However, the difference was not significant between pancreatic cancer and chronic pancreatitis except p16 gene.
Methylation of UCHL1, NPTX2, SARP2, ppENK, p16, and RASSF1A gene was detected in 6.3–37.5% of the plasma from the patients with pancreas cancer. The methylation rate of these genes from pancreatic cancer tissue was reported to be 43–100% (7 –9). The agreement degree of methylation status between tissue and plasma was highest for p16 (72.8%) and lowest for RASSF1A (9.8%). In hepatocellular carcinoma patients, the agreement of detection of p16 methylation between the plasma and tumor tissue was 73–81% (18) and it was 30% in colon cancer patients (14). This difference among the type of tumor could reflect the flow of free DNA into the plasma.
p16 gene has been reported to be methylated in 43% of the cancer tissues examined (7). In this study, p16 gene methylation was discovered in 31.3% of plasma from patients with pancreas cancer.
The method detecting methylation used in this study, MSP, is for qualitative analysis, not quantitative one. If we can use quantitative analysis for methylation such as real-time PCR, it may be possible to differentiate between pancreatic cancer and chronic pancreatitis.
It is not clear why gene methylation is observed frequently in patients with chronic pancreatitis. Chronic inflammation itself may cause methylation of genes. For example, p16INK4A gene methylation was found not only in hepatocellular carcinoma (47.8%) but also in liver cirrhosis (17.4%) (15). There is another possibility that these epigenetic changes represent a premalignant stage of chronic pancreatitis because chronic pancreatitis is one of the risk factors for pancreatic cancer. Twenty years after the diagnosis of chronic pancreatitis, the cumulative risk of pancreatic cancer was reported 4% (16). It could be feasible that precancerous of pancreatic cancer such as PanIn (8,17) might exist in the patients with chronic pancreatitis. It is expected that this question can be partially resolved through long-term follow up of patients having methylation.
There was no difference in the degree of plasma DNA methylation between cancer stage I—III and stage IV and its result coincides with previous report (18). However, others reported that methylation of RASSF1A is more common in tissue without metastasis than in metastatic cancer (7). This difference must be clarified later.
There has been two reports addressing the correlation between smoking and gene methylation in patients with head and neck cancer (19) and non-small cell lung cancer (20). In the study, there was no correlation between smoking, a risk factor for pancreatic cancer, and the degree of methylation in the plasma of patients with pancreatic cancer.
It remains unclear whether gene methylation in pancreatic cancer is related with disease prognosis. Additional studies are necessary to resolve this issue.
In summary, gene methylation was frequently detected in the plasma not only from patients with pancreatic cancer but also from chronic pancreatitis, but was rarely detected in healthy controls. Methylation status was not associated with cancer stage, tumor size, or smoking.
Footnotes
ACKNOWLEDGEMENTS
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.