
Abstract
Background
Converging evidence hints at neurodevelopmental effects in people at risk of genetic frontotemporal dementia (FTD).
Objective
We investigated total intracranial volume (TIV), a neuroimaging marker of neurodevelopment, and years of education differences between adult mutation carriers and familial non-mutation carriers, as measures of the structural and functional neurodevelopmental effects of FTD-causing genetic mutations.
Methods
This cross-sectional cohort study, facilitated through the FTD Prevention Initiative (FPI), included 902 adult pathogenic mutation carriers of GRN, MAPT, or C9orf72, and 532 familial non-carriers. ANCOVAs were computed to compare TIV and education between groups per gene. Pearson's correlations were used to examine associations between TIV and education.
Results
Mutation carriers (mean ± SD age = 50.0 ± 13.2 years, sex = 55% female, n(GRN) = 298, n(MAPT) = 187, n(C9orf72) = 417) were compared to familial non-carriers (age = 48.0 ± 12.9 years, sex = 58% female, n(GRN) = 201, n(MAPT) = 114), n(C9orf72) = 217). Consistent with prior findings in young adults, GRN carriers showed larger TIV, on average by 20531 mm3, compared to familial non-carriers (95% CI [85.4, 40977], p = 0.049, η2p = 0.008). Larger TIV correlated with higher years of education in GRN carriers (95% CI [0.01, 0.24], r(295) = 0.12, p = 0.03) and GRN non-carriers (95% CI [0.08, 0.34], r(198) = 0.21, p = 0.002). MAPT carriers demonstrated smaller TIV than non-carriers, on average by 29896 mm3 (95% CI [–58248, −1545], p = 0.039, η2p = 0.02). Models with C9orf72 and education as outcome variables did not reveal significant differences.
Conclusions
In support of the neurodevelopmental hypothesis of FTD, GRN and MAPT mutations are linked to structural neurodevelopmental changes in TIV. Further research is needed to identify mechanisms underlying neurodevelopmental influences of FTD mutations and ascertain their suitability as intervention targets.
Keywords
Introduction
Frontotemporal dementia (FTD) is a common cause of early-onset dementia.1 Its heterogenous and progressive symptomology, affecting behavior, language, and motor function, carries a devastating burden for patients and caregivers alike. 1 Genetic FTD makes up approximately 20–30% of all FTD cases, with the most common autosomal dominant mutations in one of three genes: chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and progranulin (GRN).1–4
FTD is widely recognized as a neurodegenerative disease, but various preclinical and clinical studies provide reason to suggest that there are neurodevelopmental effects of genetic FTD.5–16 The most commonly affected genes in FTD are highly penetrant and play crucial roles during neurodevelopment.5–12 There are also associations between FTD mutations and higher probability of neurodevelopmental disorders.17,18 Furthermore, a recent neuroimaging study of young adults (aged 18–29) presymptomatic for FTD found that compared to respective age-matched non-mutation carriers, C9orf72 mutation carriers had smaller total brain and thalamic volumes; MAPT mutation carriers had larger total intracranial volumes (TIV), higher levels of education, and better performance on tasks of verbal fluency and attention per the digit span forward; and GRN carriers had larger TIV and better performance on the digit symbol task. 15
Neurodevelopmental effects have been reported in other neurodegenerative diseases that have midlife onset, including genetic Alzheimer's disease 19 and Huntington's disease.20–23 In Huntington's disease, mutation carriers of pathogenic-length CAG expansions show abnormal structural, functional, and cognitive brain development compared to non-mutation carriers.20,21,23 This includes structural findings of smaller TIV 20 ; initial hypertrophy followed by declines in striatal volume in youth aged 6 to 18 years 21 ; and hyperconnectivity followed by declines in the striatal-cerebellar circuitry of youth aged 6 to 12 years, suggestive of compensatory mechanisms. 22 Functionally, improved cognitive performance was observed in young adult mutation carriers, suggestive of neurodevelopmental benefits of mutant huntingtin prior to mid-life neurodegeneration. 23
Different approaches can be used to examine neurodevelopmental markers of FTD, including neuroimaging. TIV, which includes the volume of all cranial tissues and the surrounding cerebrospinal fluid, is typically used in neuroimaging analyses to control for head size differences. However, since TIV stabilizes during adolescence, 24 around when the skull stops growing, it can serve as a proxy measure of premorbid brain growth, and thus be a plausible correlate of neurodevelopment in adult ages. 18 In normative populations, males have larger TIV than females, 25 and differences in historical year of birth, as captured by generations and likely reflecting different secular growth rates, are recognized to affect TIV measurement. 26 TIV has been employed in schizophrenia, 27 autism spectrum disorder, 28 and Huntington's 29 to examine the neurodevelopmental effects of disease.
Collectively, converging evidence hints at neurodevelopmental effects in genetic FTD.5–15,17,18 To date, only a few studies on neurodevelopment in FTD have been conducted, typically with small sample sizes, 15 and the field lacks data on youth mutation carriers and non-carriers. The present study extends the examination of TIV as a macroscopic neurodevelopmental marker in FTD in an adult sample, with the aim of identifying markers of structural and functional neurodevelopment in adult carriers of pathogenic C9orf72, MAPT, and GRN mutations. We examined TIV and years of education comparing gene mutation carriers and familial non-carriers, as measures of the potential structural and functional neurodevelopmental outcomes of genetic FTD. Consistent with prior findings 15 and the roles of C9orf72 in neurodevelopment,5,6 we hypothesized that GRN and MAPT mutation carriers would have larger TIV, and that C9orf72 mutation carriers would have smaller TIV.
Methods
Participants
This study was facilitated through the FTD Prevention Initiative (FPI). It comprised 902 adult carriers of known pathogenic mutations in GRN, MAPT, or C9orf72 (greater than 30 repeats), and 532 familial non-mutation carriers (Table 1 and Supplemental Table 1). All participants were enrolled in the Genetic Frontotemporal Dementia Initiative (GENFI; https://www.genfi.org/), or in ALLFTD (https://www.allftd.org/, NCT04363684), which combined the Advancing Research and Treatment for Frontotemporal Lobar Degeneration study (ARTFL; NCT02365922) and the Longitudinal Frontotemporal Lobar Degeneration study (LEFFTDS; NCT02372773). GENFI phase 1 and 2 included 26 clinical research centers across Europe and Canada, while ALLFTD included 18 clinical research centers across the United States and Canada.
Demographic characteristics of all participants.
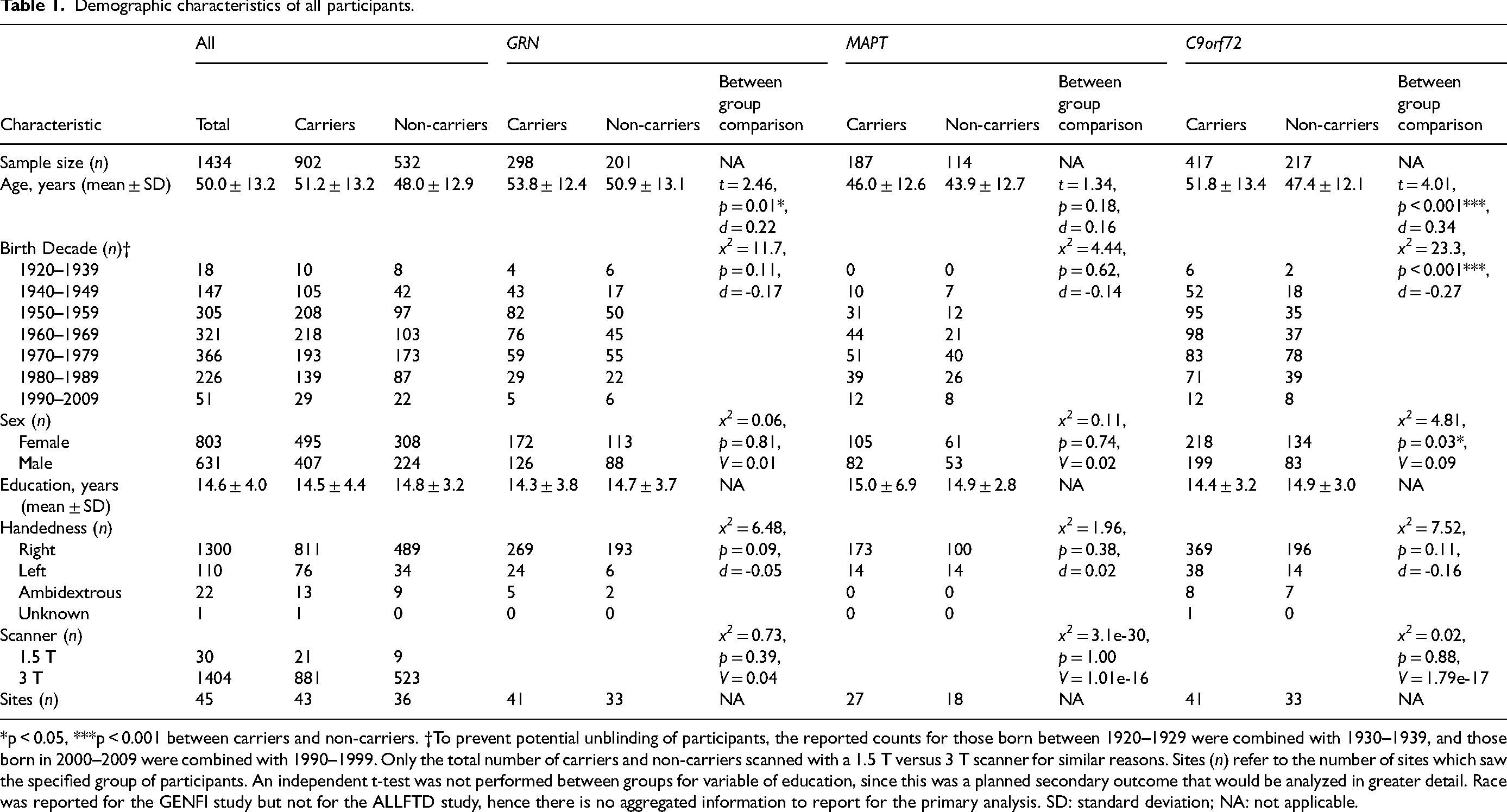
*p < 0.05, ***p < 0.001 between carriers and non-carriers. †To prevent potential unblinding of participants, the reported counts for those born between 1920–1929 were combined with 1930–1939, and those born in 2000–2009 were combined with 1990–1999. Only the total number of carriers and non-carriers scanned with a 1.5 T versus 3 T scanner for similar reasons. Sites (n) refer to the number of sites which saw the specified group of participants. An independent t-test was not performed between groups for variable of education, since this was a planned secondary outcome that would be analyzed in greater detail. Race was reported for the GENFI study but not for the ALLFTD study, hence there is no aggregated information to report for the primary analysis. SD: standard deviation; NA: not applicable.
Inclusion and exclusion criteria for the GENFI and ALLFTD studies have been described previously.30,31 Young adult GENFI participants (n = 93) who were previously reported 15 were excluded. This study included data from the following: GENFI Data Freeze 6 from Phase 1 (GENFI1, 2012–15) and Phase 2 (GENFI2, 2015–19), and ALLFTD Data Freeze 13, ARTFL/LEFFTDS from 2015–2020, and ALLFTD from 2020–2023.
Local ethics committees at each site approved the study, and all participants or their proxy decision maker provided written informed consent, in accordance with the Declaration of Helsinki.
Study design and procedures
The GENFI and ALLFTD investigations are prospective, longitudinal observational studies that collect demographic, neuroimaging, neuropsychological, behavioral, and clinical outcomes. Given the general stability of TIV and education past young adulthood, baseline demographic and neuroimaging data were analyzed in this cross-sectional cohort study. The GENFI and ALLFTD cohorts contain symptomatic C9orf72, MAPT, or GRN mutation carriers, pre-symptomatic mutation carriers, and familial non-mutation carriers. As the genes of interest in this study are highly penetrant, symptomatic and pre-symptomatic mutation carriers were considered as one group (carriers), and familial non-mutation carriers were considered the comparator (non-carriers).
Neuroimaging
GENFI and ALLFTD image acquisition and preprocessing protocols have been delineated elsewhere. 15 31–33 Pertinent to this study, GENFI participants underwent T1-weighted MRI based on the GENFI protocol using a 1.5 T (Siemens, GE) or 3 T scanner (Siemens Trio, Siemens Skyra, Siemens Prisma, Philips Achieva, GE Discovery MR750). The sequence parameters were: 256 × 256 × 208 matrix; 208 slices; 1.1 mm isotropic voxel size; flip angle of 8°; and echo time and repetition time varied by vendor. ALLFTD participants underwent T1-weighted MRI using 3 T scanners (model information reported elsewhere). 31 Magnetization Prepared Rapid Gradient Echo images were obtained using these parameters: 240 × 256 × 256 matrix; 170 slices; voxel size = 1.05 × 1.05 × 1.25 mm3; flip angle, echo time and repetition time varied by vendor.
A standard imaging protocol was used across all centers, managed, reviewed for quality, preprocessed, and had volumes extracted by the respective core imaging groups per consortia. Prior to preprocessing, all images were visually inspected for quality control, and those with excessive movements or image artifacts were removed. The preprocessing steps have been previously reported,31,33 and include bias field correction with the N3 algorithm, segmentation using SPM12 v6470 (Statistical Parametric Mapping, Wellcome Trust Centre for Neuroimaging, London, UK), normalization to a customized group template (generated using the Large Deformation Diffeomorphic Metric Mapping framework), and spatial smoothing with an 8 mm full width half maximum Gaussian kernel. TIV, which includes all gray matter, white matter, and CSF, was computed with SPM12 v6470 running under MATLAB R2014b (MathWorks, Natick, MA, USA).
Statistical analysis
All analyses were computed in R v3.6.3. Demographic comparisons between carriers and non-carriers were conducted using t-tests (age at visit) or chi-square tests (sex, birth decade, race, handedness). Statistical assumptions of data normality were determined using histograms, Q-Q plots, and the Shapiro-Wilk test (p > 0.05). Analysis of covariance (ANCOVA) assumptions of homogeneity of regression slopes, and independence of the covariates and independent variable, were also examined and passed before model computation. ANCOVAs were used to compare outcome means for the main effect of group (carriers vs. non-carriers), with separate models for the three gene groups (GRN, MAPT, C9orf72), while controlling for covariates of non-interest: birth decade, sex, and visit site. To minimize genetic heterogeneity, carrier and non-carrier comparisons were performed within each gene group rather than across pooled non-carriers. The dependent variable for the structural neurodevelopmental model was TIV and for the functional neurodevelopmental model was education; each model was computed for the GRN, MAPT, and C9orf72 groups. Visit site was used to account for unique scanners per site, and to partially account for variance in educational systems related to cultural and geographical differences. GENFI and ALLFTD participants were analyzed together in primary analyses (see Supplemental Material for findings of separate cohort analysis). Outliers for TIV and education were determined with all participant data in aggregate per gene, with a cut-off of greater than 3 SD used. Any data point classified as an outlier was removed prior to analysis.
Birth decade was included as a covariate rather than age at visit, as skull changes and TIV are understood to be stable from late adolescence. 24 Changes in secular growth rates have also been recognized to have a measurable influence on TIV changes over different generations, 26 reflecting the potential impacts of historical events on education, healthcare, nutrition, and socioeconomic conditions, which may affect neurodevelopment. 34 To capture these generational cohort effects, birth decade was coded as a categorical variable rather than a continuous measure, allowing differences between decades to be examined without imposing a linear relationship. This approach of categorizing birth by decade to control for the generational effect on changes in secular growth rate has similarly been used by others. 35 In our analysis, birth decade was determined by the year in which a participant was born; it ranged from the beginning of a decade to ten years after (e.g., 1930 to 1939, inclusive), and resulted in nine categories: 1920s, 1930s, 1940s, 1950s, 1960s, 1970s, 1980s, 1990s, and 2000s.
While there were no predictions of differential sex effects of the FTD mutations, there are known sex-specific differences in TIV 25 and education. 36 Therefore, in the primary analysis, the outcome variables of interest were analyzed with a sex-aggregated approach (males and females combined), with sex as a covariate. Planned follow-up sensitivity analyses used a sex-stratified approach, where males and females were analyzed separately. Sensitivity analysis for site was also conducted, censoring sites that had only one participant (i.e., only one carrier or non-carrier): 4/31 sites for GRN, 7/28 sites for MAPT, and 9/42 sites for C9orf72 contained only one participant. As TIV may vary by scanner field strength, sensitivity analyses for this variable were computed. For GRN and C9orf72, a separate model included scanner field strength (1.5 T versus 3 T) as an additional categorical covariate, and we compared model fit with and without scanner field strength. For MAPT, only one participant was scanned with a 1.5 T magnet, making scanner field strength collinear with other variables; therefore, we computed sensitivity analyses in two ways: (1) compared model performance when including site vs. scanner field strength as a covariate, and (2) removed the single participant scanned with a 1.5 T magnet to assess whether this impacted TIV findings.
To determine whether brain size, particularly if larger, reflects the usual positive relationship between TIV and education, per gene, Pearson's correlations were performed to examine potential associations between TIV and education for carriers and non-carriers.
Exploratory MAPT genetic mutation subtype analysis
To assess for potential differential effects of different MAPT mutations, a separate model was computed for MAPT participants with mutation type as an additional covariate. MAPT mutations were categorized into 1 of 5 types by their underlying pathophysiology and/or functional consequences, as defined previously (Supplemental Table 2). 37
Results
Participants
The primary analyses of the combined cohort examined 1434 participants, of whom 902 were mutation carriers and 532 were non-carriers (Table 1). Participants were categorized based on genetic mutations, resulting in 298 GRN carriers, 201 GRN familial non-carriers, 187 MAPT carriers, 114 MAPT familial non-carriers, 417 C9orf72 repeat expansion carriers, and 217 C9orf72 familial non-carriers. The mean age of all participants at baseline, when TIV was measured and years of education were assessed, was 50.0 years (SD = 13.2, range = 18 to 89.7).
GRN carriers were slightly older than their familial non-carriers (53.8 vs. 50.9 years, p = 0.01, d = 0.22). There were no differences in birth decade, sex, and handedness, between GRN carriers and non-carriers (p > 0.05). No differences were observed in MAPT carriers and non-carriers in any of these demographic variables (p > 0.05). C9orf72 repeat expansion carriers differed from their familial non-carriers in age (p < 0.0001, d = 0.34), birth decade (p < 0.0001, d = -0.27), and sex (p = 0.03, d = 0.19), with carriers being older, born in earlier decades, and having a higher ratio of males; however, these differences were all small in effect size (d < 0.5). No differences were observed between C9orf72 carriers and non-carriers in handedness (p > 0.05).
GRN
TIV
GRN carriers exhibited larger TIV than familial non-carriers, on average by 20531 mm3: F(1495) = 3.89, 95% confidence interval (CI) [85.4, 40977], p = 0.049, η2p = 0.008 (Figure 1(a)). As predicted, birth decade, sex, and site were associated with TIV. Greater TIV was observed in males (F(1495) = 348, p < 0.0001, η2p = 0.43) and in later birth decades (F(7495) = 2.15, p = 0.04, η2p = 0.03). There were no differences or interactions with sex in the sex-aggregated sensitivity analysis. Findings from the site sensitivity analysis, where sites containing only one participant was removed, were consistent with that of the primary analysis (see Supplemental Table 3 for outcome variable means before and after covariate adjustments, and Supplemental Table 4 for unadjusted TIV means stratified by birth decade and scanner field strength). Similarly, findings from the scanner field strength sensitivity analysis, where scanner strength was an additional model covariate, remained consistent with primary findings. There was also no difference in model fit with scanner strength included as a covariate and no significant effect of scanner strength: F(1, 456) = 0.06, p = 0.81.
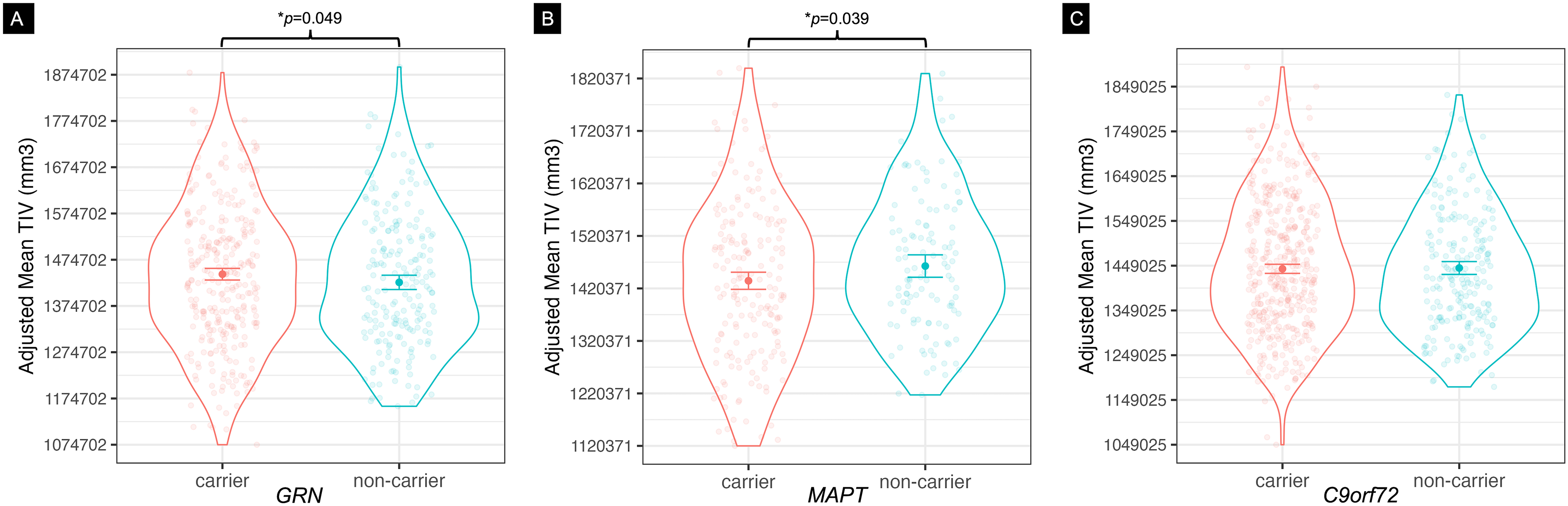
Mean total intracranial volume (TIV) for carriers and non-carriers of FTD-causing genetic mutations. TIV measured in mm3, adjusted for differences in birth decade, sex, and visit site. TIV differences were observed in (A) GRN carriers, who had larger TIV, and (B) MAPT carriers, who had smaller TIV. (C) Carriers of the C9orf72 hexanucleotide repeat expansion showed no differences in TIV relative to non-carriers. *p < 0.05.
Education. GRN carriers and non-carriers did not differ in years of education (Figure 2(a)). Main effects were observed for birth decade (F(7497) = 6.09, p < 0.0001, η2p = 8.50e-2) and site (F(30,497) = 5.06, p < 0.0001, η2p = 0.25). There were no interactions with carrier status (b = 49.4, SEM = 44.2, t(497) = 1.12, p = 0.26), and the pattern of results remained the same in the site sensitivity analyses.
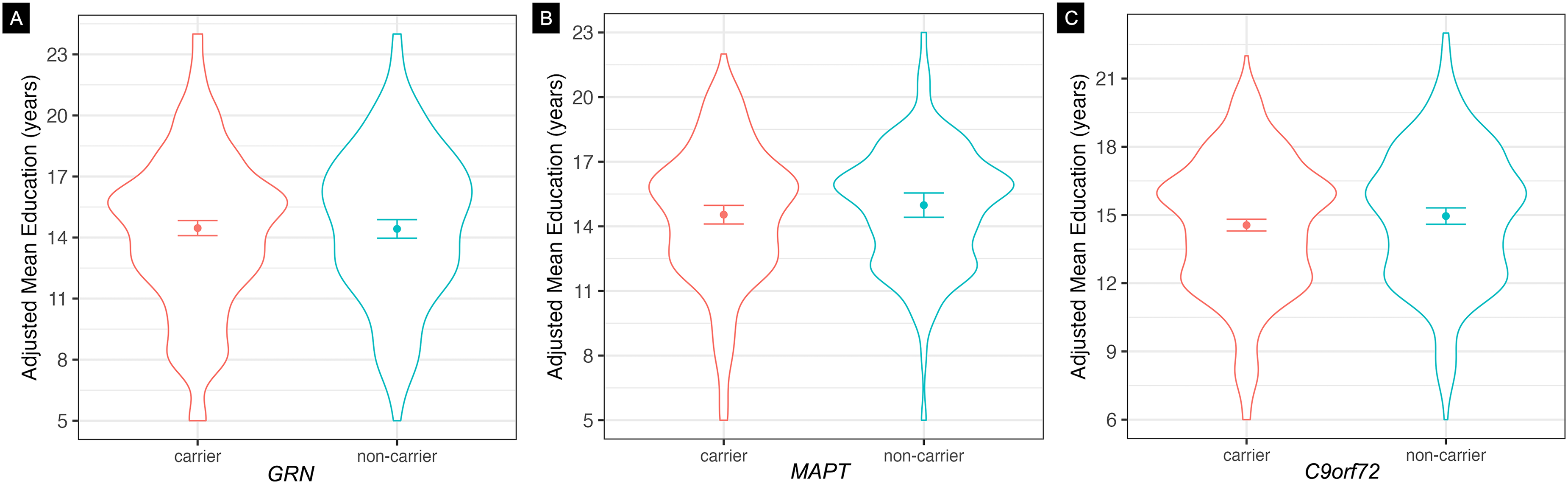
Mean years of education by carriers and non-carriers of FTD-causing genetic mutations. Measured in years, adjusted for differences in birth decade, sex, and visit site. Education differences were not observed in (A) GRN carriers, (B) MAPT carriers, or (C) C9orf72 hexanucleotide repeat expansion carriers compared to respective non-carriers.
Correlation between TIV and education
Larger TIV was associated with greater years of education in GRN carriers (95% CI [0.01, 0.24], r(295) = 0.12, p = 0.03) and non-carriers (95% CI [0.08, 0.34], r(198) = 0.21, p = 0.002).
MAPT
TIV
MAPT carriers had smaller TIV than their familial non-carriers, on average by 29896 mm3: F(1299) = 4.31, 95% CI [–58248, −1545], p = 0.039, η2p = 0.02 (Figure 1(b)). Sex was associated with TIV, with males having larger TIV: F(1299) = 192, p < 2e-16, η2p = 0.40. When sex was stratified, female MAPT carriers had smaller TIV than non-carriers: F(1163) = 4.88, p = 0.03, η2p = 0.04; no significant difference existed between groups in the male MAPT model, though the directionality of the means in male MAPT carriers and non-carriers matched that of the primary analysis and female only analysis. When MAPT mutation type was included as a covariate, there was a trend of a main effect of mutation type: F(1298) = 3.63, 95% CI [–58690, −1943], p = 0.06, η2p = 0.02, but there were no interactions with TIV. Sensitivity analyses for site, where sites with one participant were removed, yielded results consistent with the primary findings. Including scanner field strength instead of site as a covariate also produced consistent findings, and did not improve model fit: F(1, 291) = 1.33, p = 0.14. Removing the single participant who was scanned with a 1.5 T magnet resulted in near-identical findings to the primary analysis.
Education
Participants who carried a MAPT mutation did not vary in years of education compared to non-carriers (Figure 2(b)). The main effect of site was significant in its association with education (F(22,298) = 1.98, p = 0.01, η2p = 0.18), birth decade approached significance in its association with education (F(6298) = 2.06, p = 0.06, η2p = 0.04), and there was a group by birth decade interaction (b = 2.98, SEM = 1.51, t(298) = 1.98, p = 0.049); however, upon inspection, there was no consistent pattern of interest.
The removal of sites with only one participant also yielded non-significance, but separate models per GENFI and ALLFTD cohorts were interesting (Supplemental Table 5). Analysis of MAPT participants in the ALLFTD cohort alone yielded non-significance; however, in MAPT participants belonging to the GENFI cohort, the main effect of group approached significance while controlling for birth decade, sex, and site, where MAPT carriers tended to have fewer years of education than non-carriers: F(1152) = 3.46, p = 0.07, η2p = 0.03. In GENFI participants, birth decade and site also approached significance in its association with years of education: F(5152) = 2.13, p = 0.07, η2p = 0.08, and F(15,152) = 1.56, p = 0.09, η2p = 0.15, respectively. A similar pattern of results was observed when the influence of MAPT mutation type was accounted for: F(1152) = 3.83, p = 0.05, η2p = 0.03.
Correlation between TIV and education
There were no significant correlations between TIV and years of education in MAPT carriers or non-carriers. When participants were stratified by sex, the expected trend of larger TIV associated with greater years of education in male MAPT carriers was observed: 95% CI [-0.008, 0.41], r(78) = 0.21, p = 0.06.
C9orf72
TIV
There was no difference in TIV between repeat expansion carriers and non-carriers (Figure 1(c)). Main effects of sex and site were present; sex: F(1627) = 486.5, p < 0.0001, η2p = 0.46, with males having greater TIV than females; and site: F(41,627) = 2.87, p < 0.0001, η2p = 0.17. The sex and site sensitivity analyses showed similar pattern of results (Supplemental Material). Including scanner field strength as an additional covariate produced consistent findings, where no differences were observed in TIV between carriers and non-carriers (p > 0.05), but model fit was improved: F(1, 578) = 5.96, p = 0.01.
Education
No difference was observed in years of education between C9orf72 repeat expansion carriers and non-carriers (Figure 2(c)), while main effects of birth decade (F(6620) = 2.72, p = 0.01, η2p = 0.03) and site (F(41,620) = 4.52, p < 0.0001, η2p = 0.25) were observed, with no significant interactions. C9orf72 participants born in later birth decades were associated with having greater years of education. All sensitivity analyses yielded non-significant findings in similar patterns to the primary analysis.
Correlation between TIV and education
Larger TIV was linked to greater years of education in C9orf72 repeat expansion carriers (95% CI [0.02, 0.21], r(413) = 0.12, p = 0.02), and approached significance in non-carriers (95% CI [-0.02, 0.25], r(213) = 0.12, p = 0.09). When participants were stratified by sex, we observed in male C9orf72 carriers the expected trend of larger TIV correlated with greater years of education: 95% CI [0.13, 0.39], r(197) = 0.27, p = 0.0001.
Discussion
This investigation revealed that a macroscopic neurodevelopment marker, TIV, differed in GRN and MAPT mutation carriers but not in C9orf72 repeat expansion carriers relative to familial non-carriers. We did not observe differences in years of education between carriers and non-carriers of any genetic mutations of interest. Expected positive correlations between TIV and years of education were present in both carriers and non-carriers of GRN mutations or C9orf72 repeat expansions. The expected influences of sex and birth decade were observed, with males on average having larger TIV and later birth decades associated with larger TIV and greater years of education in all of the gene groups. Sensitivity analyses accounting for visit site and scanner field strength yielded results consistent with our primary findings, indicating robustness to technical and cohort-related variation. Collectively, these findings support the hypothesis that some FTD-causing mutations influence structural neurodevelopment, as measured by TIV.
Adult GRN mutation carriers have greater TIV, on average, than familial non-carriers, consistent with findings in young adults. 15 In FTD, GRN mutations are mainly heterozygous loss-of-function that lead to haploinsufficiency in progranulin, a secreted growth factor important for neurodevelopmental processes including neurite outgrowth, and neuronal differentiation and survival.2,8 Homozygous GRN mutations impair early lysosomal storage function, leading to later neurodegeneration in FTD.8,38 Progranulin also promotes synaptic development, as shown in young progranulin knockout mice that had diminished synaptic connectivity, plasticity, and dendritic spine density in the hippocampus, 9 raising the possibility that loss-of-function GRN mutations in FTD could lead to less synaptic pruning and thus greater cortical volumes and resultant larger TIV.
While there were no significant differences in education between GRN carriers and non-carriers, larger TIV correlated with greater years of education in both groups. This was expected in non-carriers, as it pertains to the usual pattern observed in healthy adults. 39 However, a similar correlation observed in GRN carriers suggests that early neurodevelopmental effects related to GRN haploinsufficiency are potentially neutral, advantageous, or compensatory with respect to brain function. This theory is supported by findings of enhanced cognitive performance in young adult GRN carriers, who performed better in the digit symbol task than non-carriers and performed as well as non-carriers across other cognitive tasks. 15 Further studies are necessary to determine the driving factors behind the increased TIV in GRN mutation carriers.
The observation of smaller TIV in MAPT carriers aligns with literature which supports links between tau and human neurodevelopment.10–12,16 For example, low plasma tau levels have been linked to adolescents with early-onset psychosis, which correlated with smaller surface area of the orbitofrontal cortex. 16 Alternative splicing of MAPT mRNA transcripts in humans has also been reported to undergo rapid and marked changes between the last trimester of fetal development and the first post-natal months, resulting in differential spatial and temporal expression of tau in the developing brain 10 ; these changes coincide with a critical phase of neuronal migration required for establishing mature neuronal connectivity. 40 In human induced pluripotent stem cell models of FTD, MAPT mutations resulted in prolonged maturation of cortical neurons and altered electrophysiological properties, 11 as well as diminished proliferation capacity of neural progenitors and disrupted Wnt/Shh signaling pathways. 12 Intriguingly, related signaling pathways have been acknowledged for regulating developmental pathways that contribute to TIV: the WNT/ß-catenin pathway is involved in meningeal development, which confers downstream effects on calvarial and brain development 40 ; and the Shh-mediated pathways regulate calvarial suture morphogenesis and osteogenesis. 41 Neurodevelopmental models will be required to determine if MAPT mutations causing FTD affect TIV via such direct mesenchymal effects, or indirect effects based on differences in maximum brain volume.
In addition, we explored the effects of MAPT mutation type, due to evidence that different MAPT mutations confer different functional consequences and underlying pathology, leading to systematic differences in age of FTD onset and eventual death. 37 Of particular interest, in induced pluripotent stem cells, the MAPT IVS10 + 16 mutation increases 4R tau isoform expression and impairs neuronal progenitor proliferation. 12 Although the effect sizes of the models marginally increased with the addition of MAPT mutation type as a covariate, there were no specific mutation type main effects or interactions.
Similar to GRN, there were no differences in years of education between MAPT carriers and non-carriers in the primary analyses. However, in a genome-wide association study, MAPT was one of the top candidate genes associated with years of education; gene function analysis revealed MAPT mutations during pre-natal stages of development to be associated with impaired dendrite morphogenesis, altered morphology of hippocampal mossy fibers, and atypical axonal guidance, 42 which are processes involved in long-term potentiation, synaptic plasticity, memory formation, and learning.
The findings of smaller TIV and no difference in years of education in MAPT carriers contrast prior findings in GENFI young adults. 15 Potential reasons for the discrepant findings include a smaller sample size in the young adult study, and birth decade or cohort effects. 15 However, we consider the present findings most robust given the larger sample size, and attribute the differences as likely due to cohort effects of the smaller sample, 15 including unmeasured differences in socioeconomic status and education systems.
Carriers of the C9orf72 hexanucleotide repeat expansion did not differ in TIV from non-carriers, consistent with prior findings. 15 Indeed, smaller gray matter volumes can occur without changes in TIV. Notably, however, the lack of TIV changes does not indicate an absence of neurodevelopmental effects. Less gyrification has been observed in asymptomatic C9orf72 repeat expansion carriers in parietal, occipital, and temporal regions 14 ; since gyrification is a process that begins during third trimester of pregnancy and peaks during childhood, these findings are supportive of neurodevelopmental consequences of C9orf72 on brain structure, even if TIV is unaffected. Others have highlighted associations between neurodevelopmental disorders and C9orf72 repeat expansion, such as a higher probability of schizophrenia and autism spectrum disorder in young relatives of C9orf72 carriers compared with non-carriers. 17 Preclinical models also support that C9orf72 affects cellular and molecular processes involved in early stages of neurodevelopment.5,6 Indeed, studies suggest that both loss-of-function 3 and gain-of-function 6 effects of C9orf72 repeat expansions contribute to neurodevelopmental effects, similar to the loss- and gain-of-function mechanisms proposed for C9orf72 induced ALS/FTD neurodegeneration. 43 These mechanisms, which engage at different times and in different brain regions in mouse neurodevelopment, 44 could counteract each other, thereby concealing macrostructural neurodevelopmental effects of C9orf72 on TIV.
C9orf72 carriers had similar years of education to non-carriers, and a normal positive correlation between TIV and years of education was observed in carriers and trending towards significance in non-carriers. We acknowledge that education levels are widely influenced by socioeconomic and environmental factors unmeasured in this cohort.45,46 Future studies including additional demographic variables known to impact education, as well as prospective cognitive assessments in youth, are needed to better delineate potential neurodevelopmental effects of C9orf72 on brain function.
While this cohort was large for a rare genetic disease and enabled high statistical power, we acknowledge several limitations. TIV models for GRN and MAPT had small effect sizes, as did the correlations between TIV and education in GRN carriers and non-carriers. Additionally, several potential confounders were not available, such as participant height, which affects cranium size and thus TIV, as well as nutritional and socioeconomic status, which influence years of education. The comparably smaller sample of MAPT participants and unequal distribution per MAPT mutation type also limited our ability to identify potential differential effects of MAPT mutations.
In summary, this study provides insight and evidence supporting the neurodevelopmental hypothesis of FTD, and has several implications for future examination. GRN and MAPT mutations are associated with TIV changes, supporting structural neurodevelopmental effects in carriers of these FTD mutations. The now replicated finding that GRN mutation carriers have larger TIV than non-carriers, and the preserved association between TIV and years of education, indicate that GRN mutations influence brain structure, and is potentially associated with advantageous functional effects during early development. Studies evaluating the cellular and molecular mechanisms by which these mutations affect development in preclinical models highlight potential mechanisms by which these FTD mutations may impact neurodevelopment. Future directions investigating these pathways in human genetic FTD models may identify novel treatment targets for maintaining function in genetic FTD beyond young adulthood.
Supplemental Material
sj-docx-1-alz-10.1177_13872877251393414 - Supplemental material for Neurodevelopmental effects of genetic frontotemporal dementia mutations revealed by total intracranial volume differences
Supplemental material, sj-docx-1-alz-10.1177_13872877251393414 for Neurodevelopmental effects of genetic frontotemporal dementia mutations revealed by total intracranial volume differences by Isis So, Arabella Bouzigues, Lucy L Russell, Phoebe H Foster, Eve Ferry-Bolder, John C van Swieten, Lize C Jiskoot, Harro Seelaar, Raquel Sanchez-Valle, Robert Laforce Jr., Caroline Graff, Daniela Galimberti, Rik Vandenberghe, Alexandre de Mendonça, Pietro Tiraboschi, Isabel Santana, Alexander Gerhard, Johannes Levin, Sandro Sorbi, Markus Otto, Florence Pasquier, Simon Ducharme, Chris R Butler, Isabelle Le Ber, Maria Carmela Tartaglia, Mario Masellis, James B Rowe, Matthis Synofzik, Fermin Moreno, Barbara Borroni, Tyler Kolander, Carly Mester, Danielle Brushaber, Kejal Kantarci, Hilary W Heuer, Leah K Forsberg, Jonathan D Rohrer, Bradley F Boeve, Adam L Boxer, Howard J Rosen, Elizabeth C Finger, and on behalf of Frontotemporal Dementia Prevention Initiative (FPI) Investigators in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
The authors thank all GENFI and ALLFTD participants and their families for their contributions to the study. Data presented in this manuscript were derived from multiple consortium comprising the FTD Prevention Initiative. The authors acknowledge the invaluable contributions of the study participants and families as well as the assistance of the support staff at each of the participating sites.
ORCID iDs
Ethical considerations
Local ethics committees at each site approved the study.
Consent to participate
All participants or their proxy decision maker provided written informed consent, in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Canadian Institutes of Health Research (CIHR) grants #470797 and #452843, held by E.F., who also holds funding from CIHR grant #327387. I.S. is supported by a BrainsCAN MD/PhD Award, CIHR Canada Graduate Scholarship-Doctoral #193336, and Parkwood Institute Research Cross-Theme Collaboration Studentship (funded by the St. Joseph’s Health Care Foundation). J.C.V.S., L.C.J. and H.S. are supported by the Dioraphte Foundation grant 09-02-03-00, Association for Frontotemporal Dementias Research Grant 2009, Netherlands Organization for Scientific Research grant HCMI 056-13-018, ZonMw Memorabel (Deltaplan Dementie, project number 733 051 042), ZonMw Onderzoeksprogramma Dementie (YOD-INCLUDED, project number10510032120002), EU Joint Programme-Neurodegenerative Disease Research-GENFI-PROX, Alzheimer Nederland and the Bluefield Project. R.S-V. is supported by Alzheimer’s Research UK Clinical Research Training Fellowship (ARUK-CRF2017B-2) and has received funding from Fundació Marató de TV3, Spain (grant no. 20143810). R.S.-V. was funded at the Hospital Clinic de Barcelona by Instituto de Salud Carlos III, Spain (grant code PI20/00448 to RSV) and Fundació Marató TV3, Spain (grant code 20143810 to RSV). RL is supported by the Canadian Institutes of Health Research and the Chaire de Recherche sur les Aphasies Primaires Progressives Fondation Famille Lemaire. C.G. received funding from EU Joint Programme-Neurodegenerative Disease Research-Prefrontals Vetenskapsrådet Dnr 529-2014-7504, EU Joint Programme-Neurodegenerative Disease Research-GENFI-PROX, Vetenskapsrådet 2019-0224, Vetenskapsrådet 2015-02926, Vetenskapsrådet 2018-02754, the Swedish FTD Inititative-Schörling Foundation, Alzheimer Foundation, Brain Foundation, Dementia Foundation and Region Stockholm ALF-project. C.G. is supported by the Swedish Frontotemporal Dementia Initiative Schörling Foundation; Vetenskapsrådet (Swedish Research Council) JPND Prefrontals, 2015–02926, 2018–02754, and JPND-GENFI-PROX 2019-02248; Swedish Alzheimer Foundation, ALF-project Region Stockholm, Karolinska Institutet Doctoral Funding, KI Strat-Neuro, Swedish Dementia Foundation, and Swedish Brain Foundation. R.V. has received funding from the Mady Browaeys Fund for Research into Frontotemporal Dementia. J.L. received funding for this work by the Deutsche Forschungsgemeinschaft German Research Foundation under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198). M.O. has received funding from Germany’s Federal Ministry of Education and Research (BMBF). S.D. receives salary funding from the Fonds de Recherche du Québec – Santé, and receives funding from the Canada First Research Excellence Fund, awarded to McGill University for the Healthy Brains, Healthy Lives initiative. M.M. was, in part, funded by the UK Medical Research Council, the Italian Ministry of Health and the Canadian Institutes of Health Research as part of a Centres of Excellence in Neurodegeneration grant, and also Canadian Institutes of Health Research operating grants (Grant #s: MOP- 371851 and PJT-175242) and funding from the Weston Brain Institute. J.B.R. has received funding from the Wellcome Trust (103838; 220258) and is supported by the Cambridge University Centre for Frontotemporal Dementia, the Medical Research Council (MC_UU_00030/14; MR/T033371/1) and the National Institute for Health Research Cambridge Biomedical Research Centre (NIHR203312). F.M. is supported by the Tau Consortium and has received funding from the Carlos III Health Institute (PI19/01637). J.D.R. is supported by the Bluefield Project and the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre, and has received funding from an MRC Clinician Scientist Fellowship (MR/M008525/1) and a Miriam Marks Brain Research UK Senior Fellowship. Several authors of this publication (J.C.V.S., M.S., R.V., A.d.M., M.O., R.V., J.D.R.) are members of the European Reference Network for Rare Neurological Diseases (ERN-RND)—Project ID No 739510. This work was also supported by the EU Joint Programme—Neurodegenerative Disease Research GENFI-PROX grant [2019-02248; to J.D.R., M.O., B.B., C.G., J.C.V.S. and M.S]. L.K.F., B.F.B., and K.K. each receive research support from the NIH. A.L.B. receives research support from the NIH, the Tau Research Consortium, the Association for Frontotemporal Degeneration, Bluefield Project to Cure Frontotemporal Dementia, Corticobasal Degeneration Solutions, the Alzheimer’s Drug Discovery Foundation and the Alzheimer’s Association. H.J.R. receives research support from the NIH and the state of California.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.D.R. has received consulting fees from UCB, AC Immune, Astex Pharmaceuticals, Biogen, Takeda, and Eisai and is part of the Medical Advisory Board for Alector, Arkuda Therapeutics, Wave Life Sciences, and Prevail Therapeutics. R.S-V. has received consulting fees from Wave Pharmaceuticals and Ionis-Biogen and from Roche Diagnostics and Janssen for educational activities and is member of the Data Safety Monitoring Board for Wave Pharmaceuticals and Ionis-Biogen. B.B. has received consulting fees from Alector and Wave Pharmaceuticals and has a pending patent on noninvasive brain stimulation. M.M. has received compensation for royalties from Henry Stewart Talks Ltd; consulting fees from Arkuda Therapeutics, Ionis, Alector, Biogen, and Wave Life Sciences; personal fees for educational activities from Alector and Arkuda Therapeutics; and travel fees from Alector Pharmaceuticals. M.C.T. has received consulting fees from Roche. C.G. has received consulting fees from Studentlitteratur 238 SEK 2020 and reimbursement for travel and lodging from University of Pennsylvania for attending KOL-meeting in October 2019. J.R. has received consulting fees from Alector, Asceneuron, Astronautx, Astex, Aviado Bio, Booster Therapeutics, ClinicalInk, Curasen Therapeutics, Cumulus Neuro, Eisai, Ferrer, SV Health, Prevail, Vesper Bio and UCB; he has provided expert testimony in private noncommercial medicolegal case reports; is part of an Advisory Board for several noncommercial academic institutions; and is Chief Scientific Advisor to Alzheimers Research UK, Guarantor of Brain, Trustee PSP Association, Trustee Darwin College, and Associate Director of the Dementias Platform UK. R.V. has received consulting fees from CyTox and is a member of the Data Safety and Monitoring Board of AC Immune. I. Santana has received consulting fees from Biogen and Roche Biogen, personal fees for presentations from Biogen, and is part of the Data Safety Monitoring Board for Novo Nordisk. S.D. has received consulting fees from Innodem Neurosciences and personal fees from Sunovion Eisai. J.L. has received consulting fees from Bayer Vital, Roche, and Biogen; is part of the Advisory Board for Axon Neurosciences; has received compensation for duty as part-time CMO from Modag; and has received author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers as well as nonfinancial support from AbbVie. M.O. has received consulting fees from Biogen, Axon, and Roche and is part of the Advisory Board for Axon. I.L.B. has received consulting fees from Alector Prevail Therapeutics and personal fees from MDS and is also part of the Data Safety Monitoring Board for Alector Prevail Therapeutics. K.K. served on the Data Safety Monitoring Board for Takeda Global Research & Development Center and data monitoring boards of Pfizer and Janssen Alzheimer Immunotherapy and received research support from Avid Radiopharmaceuticals, Eli Lilly, the Alzheimer's Drug Discovery Foundation and the NIH. B.F.B. has served as an investigator for clinical trials sponsored by Alector, Biogen, Transposon and Cognition Therapeutics; he serves on the Scientific Advisory Board of the Tau Consortium which is funded by the Rainwater Charitable Foundation. A.L.B. has served as a consultant for Aeovian, AGTC, Alector, Arkuda, Arvinas, Boehringer Ingelheim, Denali, GSK, Life Edit, Humana, Oligomerix, Oscotec, Roche, TrueBinding, Wave, Merck and received research support from Biogen, Eisai and Regeneron. H.J.R. has received research support from Biogen Pharmaceuticals, has consulting agreements with Wave Neuroscience, Ionis Pharmaceuticals, Eisai Pharmaceuticals, and Genentech. E.F. has received consulting fees for the AAN annual meeting speaker and course director honorariums; is member of the Data Safety Monitoring Board for the Lithium trial, PI E. Huey (trial funded by peer reviewed grants from ADDF); and is a scientific advisory board member for Vigil Neuroscience, Denali Therapeutics, and on an advisory panel for Biogen. M. Synofzik has received consulting fees from Janssen Pharmaceuticals, Ionis Pharmaceuticals, and Orphazyme Pharmaceuticals and received support from the Movement Disorder Society for travel.
Data availability statement
Anonymized data may be requested from the GENFI and ALLFTD projects, though certain elements of the data from both consortia may be restricted to protect the confidentiality of participants. Analytic R code can be made available upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.