
Abstract
Background
Drug repurposing offers a rapid, cost-effective approach for discovering therapies against multiple targets.
Objective
Here, we screen virtual ligand libraries consisting of 3468 approved drugs against 11 protein targets associated with Alzheimer's disease (AD).
Methods
We employ blind molecular docking, and target amyloid-β (Aβ), microtubule-associated protein tau (MAPT), Apolipoprotein E4 (APOE4), acetylcholinesterase (AChE), butyrylcholinesterase (BChE), amyloid-β protein precursor (AβPP), β-secretase (BACE1), brain-derived neurotrophic factor (BDNF), presenilin 1 (PSEN1) and 2 (PSEN2), and α-synuclein (SNCA) proteins using AutoDock Vina.
Results
Notably, multitarget binding recurs among the top-10 ligands with Ergotamine and Dihydroergotamine potentially binding 8; Dutasteride 7; Drospirenone and Nilotinib 6; Adapalene and Conivaptan 5; Bromocriptine 4; and Rolapitant, Irinotecan, Plerixafor, Saquinavir, and Telmisartan 3, out of 11 protein targets. As such, we reveal potential binding sites for ergot alkaloids, steroids, retinoids, antivirals, angiotensin receptor blockers, and Neurokinin 1 (NK1) receptor antagonists on multiple AD targets. Importantly, the therapeutic potential of the top-scoring ligands is confounded by pharmacokinetics and adverse-effects. For example, poor blood-brain barrier (BBB) penetration, and vasoconstriction, discount ergot-alkaloid use in AD. Likewise, potential toxicity limits prolonged use of steroids, Nilotinib, Adapalene, and Irinotecan. Conversely, BBB penetration, neuronal protection, oral availability, anti-inflammation, and anti-hypertension, admit Angiotensin receptor blockers (ARB), (Telmisartan/Candesartan); Antidiuretic hormone (ADH) inhibitors (Conivaptan/Tolvaptan); and of the NK1 receptors antagonists (Rolapitant/Netupitant) use in AD.
Conclusions
Our multitarget screening identifies selective synergistic AD modulators, such as ARB, ADH and NK1 receptor inhibitors, and simplifies drug discovery by focusing on the most promising candidates for experimental validation.
Keywords
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by the accumulation of amyloid-β (Aβ) and tau proteins in the brain. 1 AD is the most common chronic neurodegenerative disease worldwide, and the fifth leading cause of death in the adult population. 2 Despite the significant public health threat posed by AD, current therapeutic options are limited. Only a handful of drugs are approved by the Food and Drug Administration (FDA), and their efficacy primarily focuses on managing symptoms, rather than halting disease progression. 3 Notably, treatment of comorbid conditions associated with AD has been linked with lower disease prevalence over the last decade. 4 Comorbid conditions linked with AD include hypertension, pain, cardiovascular disease, dyslipidemia, atherosclerosis, and type 2 diabetes. 5 Addressing these conditions has been shown to significantly improve the proteopathic hallmarks of AD.
Multiple proteins have been linked to AD pathology, and promote the complex etiology and progression of the disease. 6 For example, Aβ peptides, central to the amyloid cascade hypothesis, are derived from the amyloid-β protein precursor (AβPP), through sequential cleavage by β-secretase (BACE1), and γ-secretase, the latter comprising presenilin 1 (PSEN1) and 2 (PSEN2) subunits. 7 Oligomerization of Aβ is thought to initiate a cascade of events leading to neuronal dysfunction and death. 8 In addition, microtubule-associated protein tau (MAPT), undergoes abnormal hyperphosphorylation in AD, leading to its dissociation from microtubules, and aggregation into neurofibrillary tangles. 9 Aggregation of Aβ and tau has also been linked with depletion of brain-derived neurotrophic factor (BDNF), a protein associated with synaptic plasticity in learning and memory. 10 One of the strongest risk factors of AD, is Apolipoprotein E4 (APOE4), an allele believed to promote BBB dysfunction, Aβ aggregation, and tau phosphorylation.11,12 In turn, Aβ accumulation, tau hyperphosphorylation, and APOE4 allele have been linked with high levels of the presynaptic protein α-synuclein (SNCA) in the cerebrospinal fluid (CSF). 13 Finally, acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) are reported to promote the formation of amyloid fibrils and tau hyperphosphorylation. 14 These proteins offer a rich array of targets for screening potential AD drugs.
Computational methods play a key role in modern drug discovery and significantly accelerate the process by providing powerful tools to enhance various stages. They enable virtual high-throughput screening of chemical libraries, significantly reducing the time and cost associated with experimental screening. 15 Virtual screening along with computational ADME evaluation techniques helps prioritize compounds for experimental testing. In the past, we have used such techniques to identify potential MCL1 inhibitors in a virtual library containing FDA-approved drugs. 16 Notably, these predictions were confirmed experimentally in a later study aimed at drug repurposing. 17
Drug repurposing is an innovative strategy that identifies new therapeutic applications for existing drugs. This approach offers several advantages over traditional drug discovery as it reduces time and cost of drug development process. More importantly, the approach reduces the risk of drug failure in later clinical stages as FDA-approved drugs being repurposed have already passed safety and toxicity studies. 18 To further increase success rate, multitarget virtual screening approaches have been developed. For example, Iqbal et al. neatly identified natural-like compounds that bind multiple protein targets associated with AD such as BACE, AChE, BChE MAOs, GSK3β, and NMDA.19–21 This approach supports our notion that AD is a spectrum of disorders rather than a single disease. 22 This approach has also led to several studies targeting proteins less commonly associated with AD, such as ROCK2, 23 CALHM1, 24 and Arginase. 25 Recently, two papers describing the potential repurposing of the FDA-approved drugs ergotamine, dihydroergotamine, and bromocriptine for the treatment of AD were published.26,27 Unfortunately, these publications ignore the adverse-effects of ergot alkaloids, such as vasoconstriction, and their potential aggravation of hypertension, a frequent AD comorbidity. The studies even neglect basic pharmacological properties such as blood-brain barrier (BBB) penetration. These studies encouraged us to perform our own analysis.
Here, we virtually screen for multitarget drugs with repurposing potential in the treatment of AD. In particular, we use molecular docking to identify ligands in approved drug libraries that potentially bind multiple AD protein-targets.
Methods
Ligand preparation
To prepare virtual ligand libraries for molecular docking, a dataset containing 1379 drugs approved by the FDA was downloaded from the Zinc database (zinc.docking.org/substances/subsets/fda). A separate dataset including 2068 drugs approved by other drug-agencies of the world was also downloaded (zinc.docking.org/substances/subsets/world-not-fda). Taken together, these libraries encompassed 3447 drugs currently approved worldwide. All library ligands were prepared following standard protocols, and involved adding polar hydrogens, optimizing geometry, assigning rotatable bonds, adding Gasteiger charges, and converting the files to PDBQT format for virtual screening with AutoDock Vina. 28 Ligand preparation was performed using Open Babel 3.1.1 with a Python script to automate the process. 29
Target preparation
To prepare targets for molecular docking, the 3D structures of 11 proteins associated with AD were obtained from the Protein Data Bank (https://www.rcsb.org/). These proteins included Aβ (PDB ID 2LFM:A), AChE (PDB ID 1C2B:A), APOE4 (PDB ID 6NCO:A), AβPP (PDB ID 2FKL:A and 2FJZ:A), BACE (PDB ID 2G94:A), BChE (PDB ID 4TPK:A), BDNF (PDB ID 1BND:A), MAPT (PDB ID 5E2W:P), PSEN1 (PDB ID 7Y5T:B), PSEN2 (PDB ID 7Y5X:B) and SNCA (PDB ID 1XQ8:A). All protein targets contained the human sequence of amino acid, except for AChE which contained that of Electrophorus electricus with 90% and 94% sequence identity and homology, respectively. To remove native ligands, non-standard residues, and non-target chains, visualization tools such as PyMol(Schrödinger), and ChimeraX were used. 30 To remove water molecules, add polar hydrogens and Kollman charges, and convert the files to PDBQT formats, AutoDockTools 1.5.7 was utilized. 28
Grid-box settings
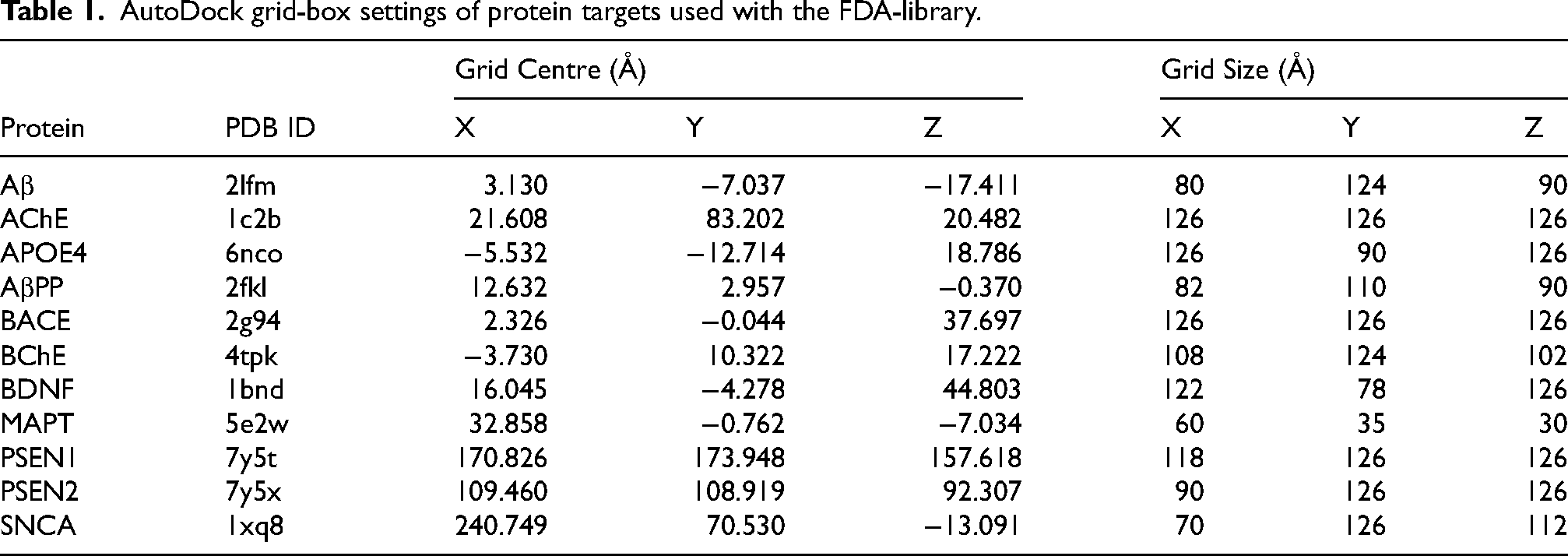
To support blind docking, the entire volume of the target protein was encompassed within the grid-box, with an additional 8–10 Å added in all directions. This measure accounted for potential ligand binding anywhere on the protein surface, or within it. Table 1 summarizes the grid-box parameters used with AutoDockTools 1.5.7 for virtual screening with the FDA-library. Similarly, Supplemental Table 1 shows the grid-box parameters used for virtual screening with the world-library. The two grid-boxes were centered slightly differently, but both encompassed the entire protein volume for blind docking.
AutoDock grid-box settings of protein targets used with the FDA-library.
Molecular docking
To virtually screen ligand-libraries against AD protein-targets, molecular docking was performed with AutoDock Vina 1.1.2. 28 To automate the screening process, an in-house Python script was employed. All prepared ligands were docked against the 11 AD protein targets, and between 9 and 10 poses were calculated. For each ligand, the pose with the lowest binding energy was picked as the best bound ligand. For each target, the 10 best bound ligands, with the lowest binding energy, were taken as the top-10 ligands (of a target). To compare the binding sites and modes of interaction, the top 10 ligands were superimposed and visualized with PyMol. The top 10 ligands were screened for recurrence across targets, and the most frequently occurring ligands, that target multiple proteins, were considered multitarget ligands.
Heatmap analysis
To identify drug sets targeting the exact same combination of proteins, we performed a heatmap analysis on the multitarget ligands, focusing on protein sets that included at least 3 proteins, and shared at least 2 drugs. This classified the top drugs that target the same subset of proteins, and vice versa.
Interaction analysis
To better understand the binding mechanism of drug-ligands to target-proteins, we employed the Protein-Ligand Interaction Profiler (PLIP), 31 an automated web-based tool (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index). PLIP analyzes protein-ligand interactions based on the PDB file format of protein-ligand complexes generated using PyMol. The tool identified and characterized the interaction types as well as the involved residues.
MD simulations
Heavy-atom MD simulation of protein-ligand complex of AChE (PDB ID 1C2B) with 13 top drug ligands was performed with NAMD (Nanoscale Molecular Dynamics program; version 3.0.1) 32 using CHARMM36 forcefield (toppar_c36_jul19.tgz). 33 To generate protein parameter files and ligand parameter files VMD (Visual molecular dynamics; version 2.0.0), 34 and CHARMM-GUI Ligand Reader and Modeler module 35 were used, respectively. Solvation and ionization of the protein-ligand complex were performed using VMD, and a standard TIP3 water model was added as with periodic boundary conditions, padded by 10 Å in each dimension. 36 Na + and Cl− ions were used to neutralize the charge. The CgenFF version of CHARMM-GUI 37 was used to generate topology and parameter files for the 13 ligands. The particle-mesh Ewald method was used for electrostatic interactions of the system. 38 Minimization was performed for 1000 steps at a constant temperature of 298 K, and production was run using NPT (constant particle number, pressure, and temperature). Thirteen sets of simulations were performed for 50 ns each. RMSD trajectory plugin tool in VMD was used to calculate the RMSD of protein and ligand.
Pharmacokinetic analysis
To evaluate the pharmacological potential of drugs in the treatment of AD, the ability to penetrate the BBB is critical. Therefore, we predicted the BBB permeability, in addition to other parameters, such as volume of distribution at steady state (VDSS), and maximum daily tolerated dose (MTD), for select top multitarget ligands using pqCSM with their SMILES annotation (https://biosig.lab.uq.edu.au/pkcsm/prediction). 39
Results
Molecular docking
In this study, we screen two virtual libraries corresponding to 1379 and 2068 drugs, approved by the FDA, and by other agencies of the world, respectively (Total of 3447 drugs). Ligand preparation increases the number of ligand isoforms to 1400 and to 2115 in the FDA and world-library respectively (Total of 3515 ligands). Ligands are screened using blind molecular docking against 11 protein-targets highly associated with AD, namely Aβ, AChE, APOE4, AβPP, BACE, BChE, BDNF, MAPT, PSEN1, PSEN2, and SNCA. Figure 1 shows the top 10 ligands of the FDA-library docked to 11 protein targets, with the binding energies obtained from AutoDock Vina, and the chemical structures of each ligand. (Supplemental Figure 2 shows the top 10 ligands of the world-library.) Notably, our blind docking simulations result in ligand clusters located in the major protein ligand-binding site. The consistent ligand localization across most proteins underscores the robustness of the docking simulations and highlights the presence of well-defined and energetically favorable binding pockets. Notable exceptions are observed for protein targets, such as AβPP which demonstrate a contiguous diverging binding groove across its surface, and PSEN1 and PSEN2 which exhibit 3 major binding regions with distinct binding sites. This divergence suggests the presence of multiple druggable pockets within these proteins.
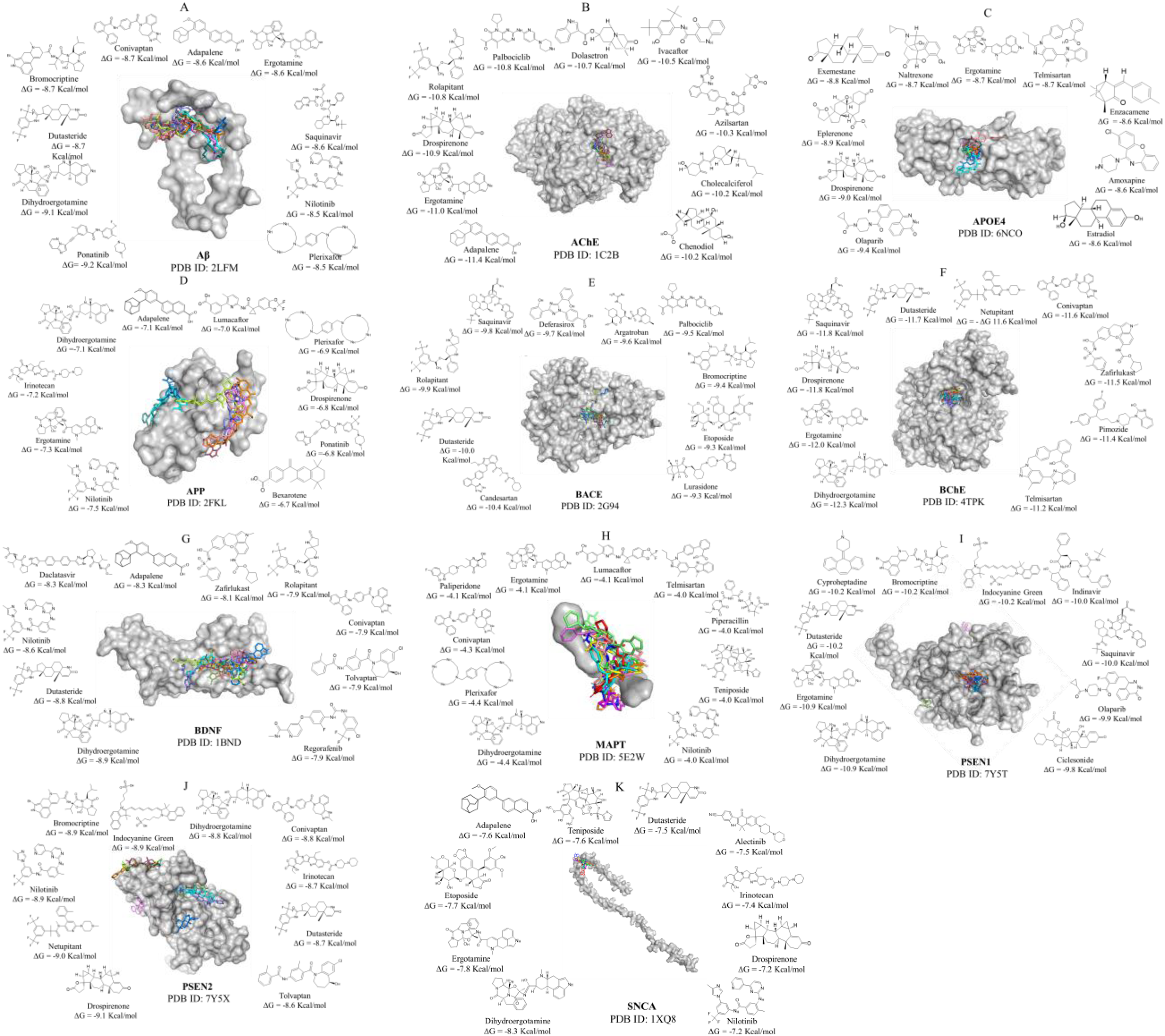
Molecular docking of FDA-approved drug-ligands onto protein-targets linked with Alzheimer's disease (AD). Shown are the top 10 ligands docked to (A) Aβ (B) AChE (C) APOE4, (D) AβPP, (E) BACE, (F) BChE, (G) BDNF, (H) MAPT, (I) PSEN1, (J) PSEN2 and (K) SNCA. For each complex, the protein-target is represented as a grey surface, and the drug-ligands are represented as sticks. Around each complex is a clockwise display of the top 10 drug-ligands ordered by binding affinity. The display includes chemical structure, generic name, and binding energy of each drug ligand. The figure was prepared using PyMol (Schrödinger).
The top 10 ligands of the cholinesterases potentially bind very strongly to AChE (ΔG between −11.4 and −10.2 kcal/mol) and BChE (ΔG between −12.3 and −11.2 kcal/mol). The drugs bind at an allosteric site, at the opening of the gorge leading into the catalytic site (Supplemental Figure 1, A2, A5). The drugs occlude access to the active site and prevents catalytic breakdown of the native ligand acetylcholine, thus potentially reducing cholinergic dysfunction associated with AD.40,41 The top 10 ligands of APOE4 potentially form strong interactions with the protein (ΔG between −9.4 and −8.6 kcal/mol) inside a pocket lined with hydrophilic residues W34 and L149 of the LDL receptor binding region (Supplemental Figure 1, A3, D2, F1). The drugs occupy the same pocket as another APOE4 stabilizer, which was designed using fragment-based discovery. 42 As such, the top 10 drugs could stabilize the APOE4 allele variant, which has the most damaging effects on neurons. 43 The top 10 ligands of BACE1 potentially bind strongly to the protein (ΔG between −10.4 and −9.3 kcal/mol), and form electrostatic interactions and hydrogen bonds with residues Y71, Q73, I105, and I118, at the entrance of the active site domain (Supplemental Figure 1, C2). As such, the drugs prevent access of native ligands to catalytic site residues D32 and D228 of BACE1, and potentially prevent AβPP cleavage by β-secretase. 44 The top 10 ligands of PSEN1 potentially bind strongly to the protein (ΔG between −10.9 and −9.8 kcal/mol) and form interactions with residues lining the active site pocket, thus inhibiting catalysis of native ligands, except for Indocyanine Green, Olaparib, and Ciclesonide. For instance, Dutasteride, Ergotamine, and Dihydroergotamine form interactions with V77, V272, I282 (Supplemental Figure 1, C4, B4, A6), and thus potentially allosterically prevent AβPP cleavage by γ-secretase. 45 The top 10 ligands of PSEN2 potentially bind strongly to the protein (ΔG between −9.8 and −8.6 kcal/mol), and form interactions with residues lining the active site pocket for native ligands, except for Indocyanine Green, Netupitant, and Tolvaptan. For example, Dutasteride, Drospirenone, and Dihydroergotamine form interactions residues, L179 and I235 (Supplemental Figure 1, B5, C5, D5) and potentially prevent AβPP cleavage by γ-secretase. 46 The fact that Bromocriptine inhibits both PSEN1 and PSEN2, the major components of γ-secretase, agrees with earlier experiments showing that Bromocriptine reduces amyloid-β production in cell lines. 47 The top 10 ligands of SNCA potentially bind to the protein (ΔG between −8.3 and 7.2 kcal/mol), and form multiple interactions with α-synuclein. The native monomeric form of α-synuclein is intrinsically disordered, and the ligands potentially stabilize monomeric conformations less prone to aggregation. While α-synuclein is more associated to Parkinson's disease, than to AD, it also co-aggregates with Aβ. 48 The drugs bind to α-synuclein, in various positions and potentially solubilize it, and prevent aggregation. The top 10 ligands of BDNF potentially binds near the interface of its dimeric form (ΔG between −8.9 and −7.9 kcal/mol), and potentially stabilize this construct, which binds to TrkB tyrosine kinase receptor, to improve neuronal plasticity. 49 The ligands do not bind to BDNF domain interacting with its receptor, and potentially do not interfere with beneficial receptor activation. The top 10 ligands of MAPT potentially bind weakly to the peptide (ΔG between −4.4 and −4.0 kcal/mol) and form interactions with the phosphorylated residues of MAPT, potentially stabilizing the monomeric state and preventing tau aggregation. 50 The top 10 ligands of AβPP potentially bind weakly (ΔG between −7.5 and −6.7 kcal/mol) to this copper-binding domain, consisting of residues 133–189. 51 The top 10 ligands insert into a contiguous curved groove on its surface. At one end of the binding groove, Dihydroergotamine and Drospirenone form interactions with residues V129, and F135 (Supplemental Figure 1, B2 and D2). At another end of the binding groove, Ergotamine forms contacts with D167 and M170 (Supplemental Figure 1, A4). These binding modes potentially stabilize the native isoform of the copper-binding domain of AβPP and potentially prevent unfolding and cleavage. The low-range affinity of ligands bound to MAPT and AβPP, express their low significance in our study of druggable AD targets.
In the past, our docking methodology has been validated through high correlation between predicted and experimental binding energies.16,52,53 Here too, predicted and experimental binding energies are closely related. For example, the experimental binding constant of irinotecan to AChE is Ki = 51 nM. 54 Using the equation Gibbs free energy equation ΔG° = −RT ln K, with a temperature of 298°K, and the gas constant 1.9872 × 10−3, the experimental binding energy corresponds to ΔG°Experimental = −9.77 Kcal/mol. Remarkably, the experimental binding energy is close to that predicted by Vina, ΔG°Predicted = −9.6 Kcal/mol. The close relationship between predicted and experimental binding energy attests to the strength of our molecular docking methodology.
Frequency analysis
A histogram of the top 10 FDA-ligands is shown in Figure 2. Notably, the top 10 ligands exhibit a significant trend towards multitarget interactions. Twenty-three ligands target 2 or more proteins (Figure 2A), while thirteen ligands target 3 or more proteins (Figure 2B). Collectively, these thirteen ligands target all the 11 proteins studied. Among the multitarget ligands, two of them, Ergotamine and Dihydroergotamine, each target 8 out of the 11 proteins. Together, the pair targets 10 of the 11 proteins, demonstrating a remarkable versatility in their binding capabilities. Following closely behind is Dutasteride, targeting 7 proteins, then Drospirenone and Nilotinib, each targeting 6 proteins, and then Adapalene and Conivaptan, each targeting 5 proteins. The multitarget ligand similarities is remarkable and reveals a common denominator previously unknown. The multitarget ligand similarities suggest either: (A) A convergent evolution of 11 protein targets associated with AD. Or (B) An almost complete lack of selectivity of ligands recurring in 4 or more targets (i.e., false positives). Option B is more plausible than option A based on the principle of Occam's razor, and the dilemma awaits experimental resolution. Consequently, the multitarget ligands are double-edged swords, with the potential to act in synergy, or in complete disarray; and to either drive, or mitigate AD progression.
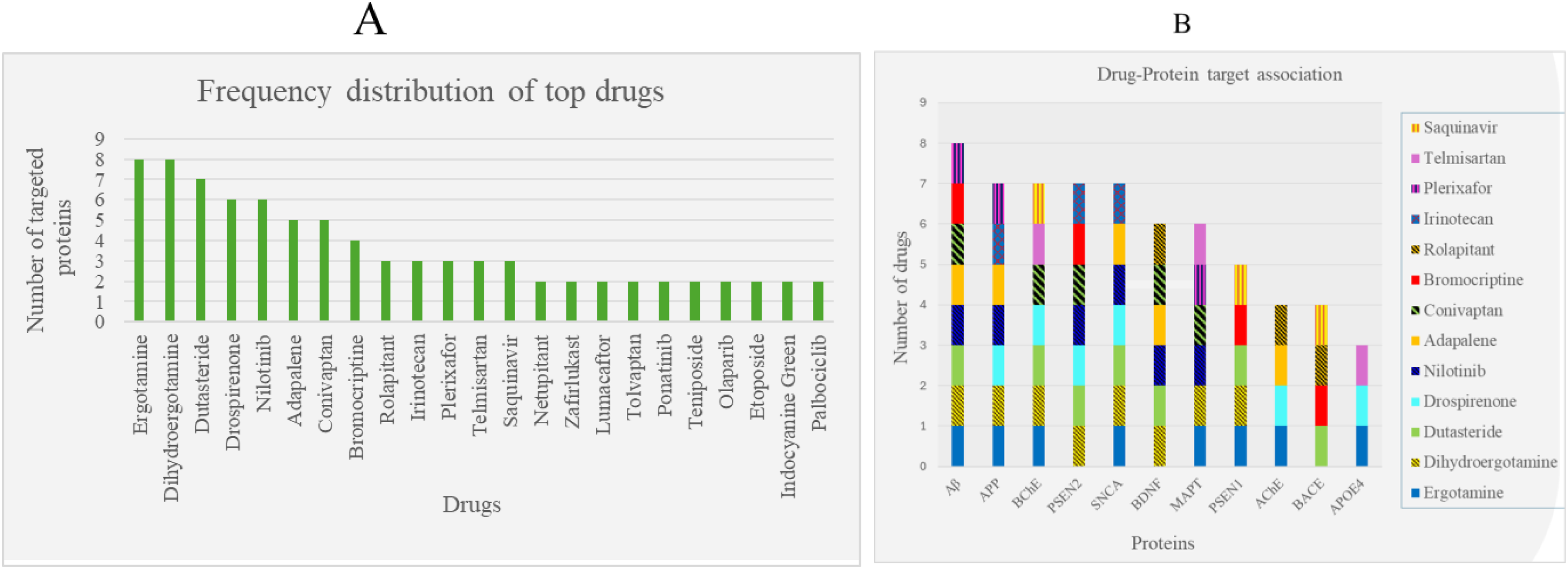
Histograms of top 10 drugs targeting proteins linked to Alzheimer’s disease (AD). Shown are (A) the number of proteins targeted by 23 of the top 10 drugs, and (B) the number of top 10 drug-ligands accommodated by the 11 protein-targets. Included in A are only 23 drugs that target two or more proteins, and in B only 13 drugs that target three or more proteins.
Interestingly, drugs that target Aβ are also most recurrent, and consist of 8 multitarget ligands shared with multiple protein targets (Figure 2B). This highlights the importance of Aβ as a representative AD target, and potentially suggests that drugs bound to Aβ, also bind other AD proteins. Similarly, drugs that target AβPP, BChE, PSEN2, and SNCA, consist of 7 multitarget ligands shared with several other proteins (Figure 2B). AβPP shares 7 of its top 10 ligands with other protein-targets, but the absence of a well-defined non-contiguous binding site, and its average affinity for ligands preclude any meaningful binding (Figure 2B). As such, we consider Ergotamine and Dihydroergotamine, drugs bound to AβPP as unselective due to their low binding affinity. Both target AβPP and interact with most AD proteins, including those with the fewest common drugs, such as APOE4 and BACE (Figure 2B). This highlights the ergot alkaloids’ structure as the most common denominator shared by the targets, and the least selective multitarget drugs among the top 10 ligands. PSEN2, a subunit of the γ-secretase complex responsible for the final cleavage of AβPP to produce Aβ, was also among the most frequently targeted proteins, but ligands bind with high affinity, and in one 3 well defined binding sites. A significant number of top 10 ligands also strongly target BChE, responsible for the breakdown of acetylcholine, a neurotransmitter central to the cholinergic dysfunction hypothesis. 55 Cholinesterase inhibition is expected to compensate for the cholinergic dysfunction by rescuing available acetylcholine. 56 Finally, APOE4, widely recognized as the strongest genetic risk factor for AD, 11 was also targeted by Ergotamine, an ergot alkaloid. Our multi-target approach is particularly relevant in a complex disorders like AD, where multiple pathways can co-contribute to disease progression.
Heatmap analysis
To better understand the interaction patterns of protein-targets and drug-ligands, a heatmap analysis is rendered in Figure 3. To screen for significant patterns, the heatmap includes only 11 proteins sharing 2 or more drugs, and 13 drugs targeting 3 or more proteins. Accordingly, the heatmap identifies robust multi-target interaction patterns, and produces a total of 26 union-sets, of 3, 4, 5, or 6 proteins sharing 2, 3 or 4 drugs. Of the 26 protein-sets, 3 union sets contain 6 proteins that share 2 drugs, namely: (1) Ergotamine and Dihydroergotamine, (2) Nilotinib Dihydroergotamine, and (3) Dutasteride and Dihydroergotamine. Following closely behind are 2 union sets containing 5 proteins that share 2 drugs; 9 union sets comprising 4 proteins that share 2–3 drugs; and 12 union sets containing 3 proteins that share 2–4 drugs.
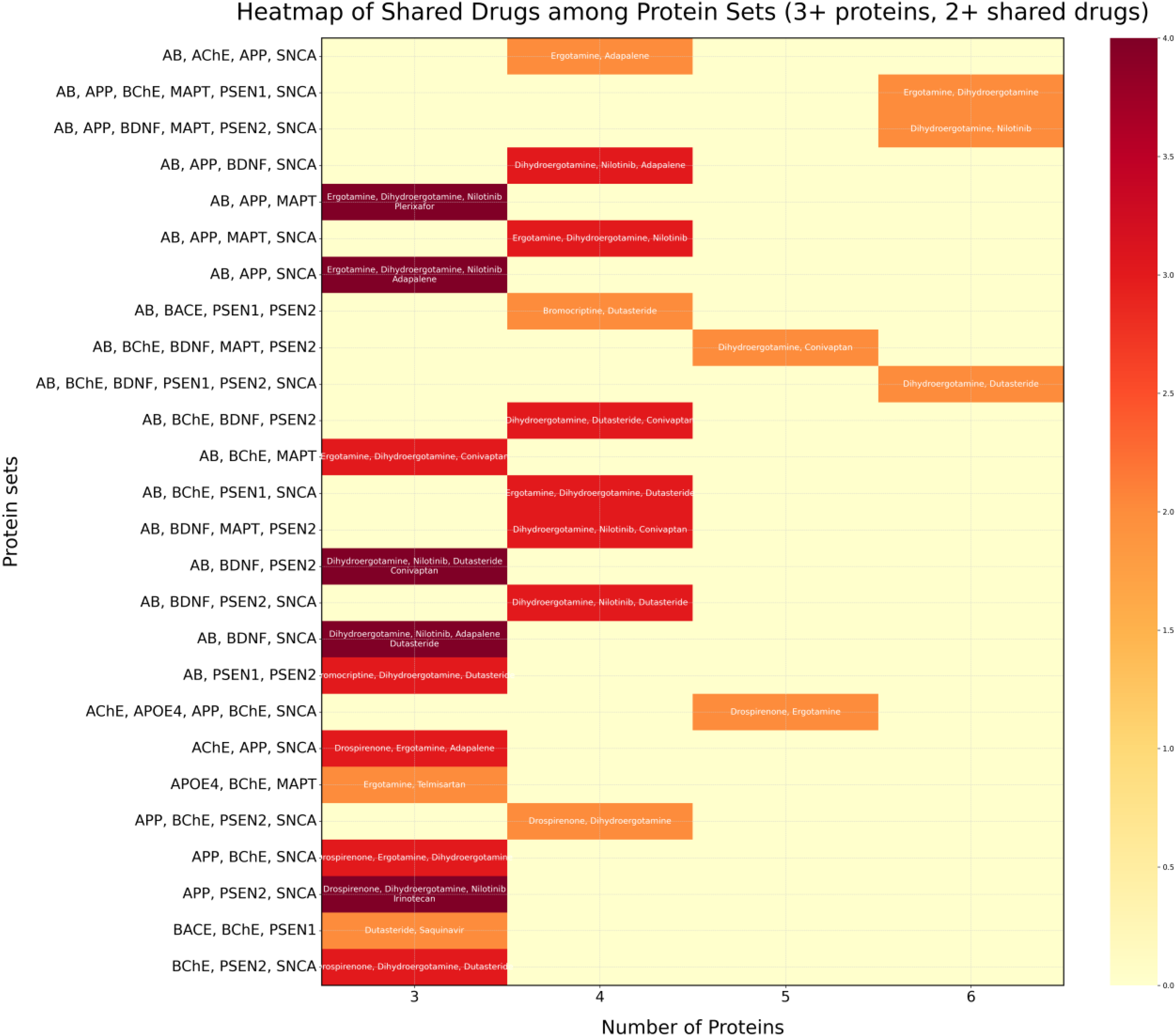
Heatmap of interactions. Shown are the union sets of proteins sharing bound drugs. The number of shared drugs is indicated by the red color intensity.
Interaction analysis
To analyze the structural differences among the best hits, a drug-protein interaction analysis is shown in Supplemental Figure 1 for selected targets and top-ligands to understand the contributing factors to binding energy. These interactions are important and provide insight into the binding mechanism. Various interactions are identified, including hydrogen-bonds, salt bridge, π-cation, π-stacking, and hydrophobic interactions. The drugs show higher binding affinity (i.e., lower binding energy) for the cholinesterases than for other protein targets. The high affinity associated with cholinesterase ligands is primarily due to the deep gorge of the enzyme active site, lined with aromatic residues which form an electrostatic potential pit with multiple binding interactions. BChE has a larger active site gorge, resulting in a more accommodating binding site. As shown Supplemental Figure 1 G4, the interaction profile of Drospirenone with PSEN1 and PSEN2 shows a high variety of interactions, both hydrogen-bonds and hydrophobic interactions. These examples clearly illustrate the contribution of buried hydrophobic surface to binding energy. This is also shown in the case of Dutasteride's equal affinity for BACE and PSEN1 (Supplemental Figure 1 G3), with an even number of hydrophobic interactions, but uneven amount of hydrogen bond, and salt bridges.
MD simulations
Molecular dynamics simulations up to 50 ns were carried out, to assay the top drug-ligand interactions with AChE. Hydrogen bond formation, solvent accessible surface, gyration radius, RMSD, and RMSF analyses of protein-ligand complexes were assessed to evaluate stability (Figure 4 and Supplemental Figure 3). Remarkably, all the ligands, Ergotamine, Dihydroergotamine, Dutasteride, Drospirenone, Nilotinib, Adapalene, Conivaptan, Bromocriptine, Rolapitant, Irinotecan, Plerixafor, Saquinavir, and Telmisartan remained in their binding sites for 50 ns. Remarkably, the top AChE ligands, Adapalene, Ergotamine, Drospirenone, Rolapitant, and Dutasteride, deviated less than 2 Angstrom in the binding site for the entire length of the simulation (50 ns). In contrast, Bromocriptine exhibited the most variation in the AChE binding site. Thus, our predictions should be taken with a grain of salt, and they require experimental validation.
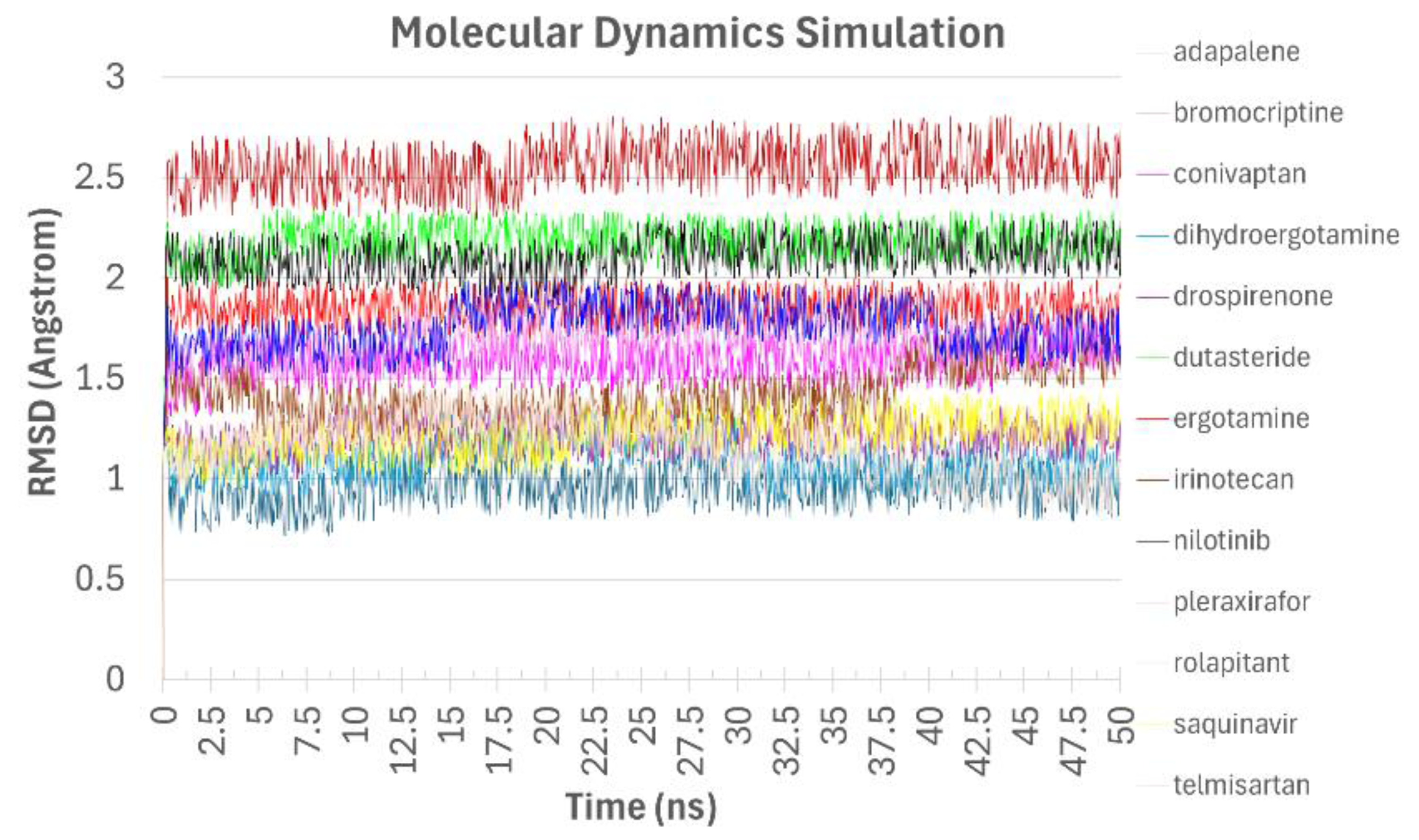
MD simulation of drugs bound to AChE. Shown are RMSD values of the top 13 drugs bound to AChE during the MD simulations. Ligand RMSD is displayed using different colors.
Pharmacokinetics
The primary goal of pharmacology is to simultaneously obtain a maximal therapeutic effect, and a minimal toxic effect. Predicted pharmacokinetics are shown in Table 2, and include BBB permeability, MTD, and VDSS. The predicted pharmacokinetic values are based on theoretical calculations, and do not replace experimental values. The predicted values are shown solely for easy comparison. BBB permeability is a measure of brain absorption, and is of great importance in AD. A positive LogBB value indicates good BBB penetration, while a negative one suggests poor brain accessibility. The MTD is another measure of safety. High MTD values indicate that high drug doses are safe and can help achieve therapeutic concentrations at the brain target site, in case of low BBB permeability. Finally, the VDSS is a measure of drug distribution. Low VDSS is indicative of strong plasma binding, while high VDSS suggests extensive distribution into tissues. For drugs with low BBB penetration, low VDSS can help maintain higher plasma concentrations, potentially enhancing delivery to the brain.
Predicted pharmacokinetics of top drugs.
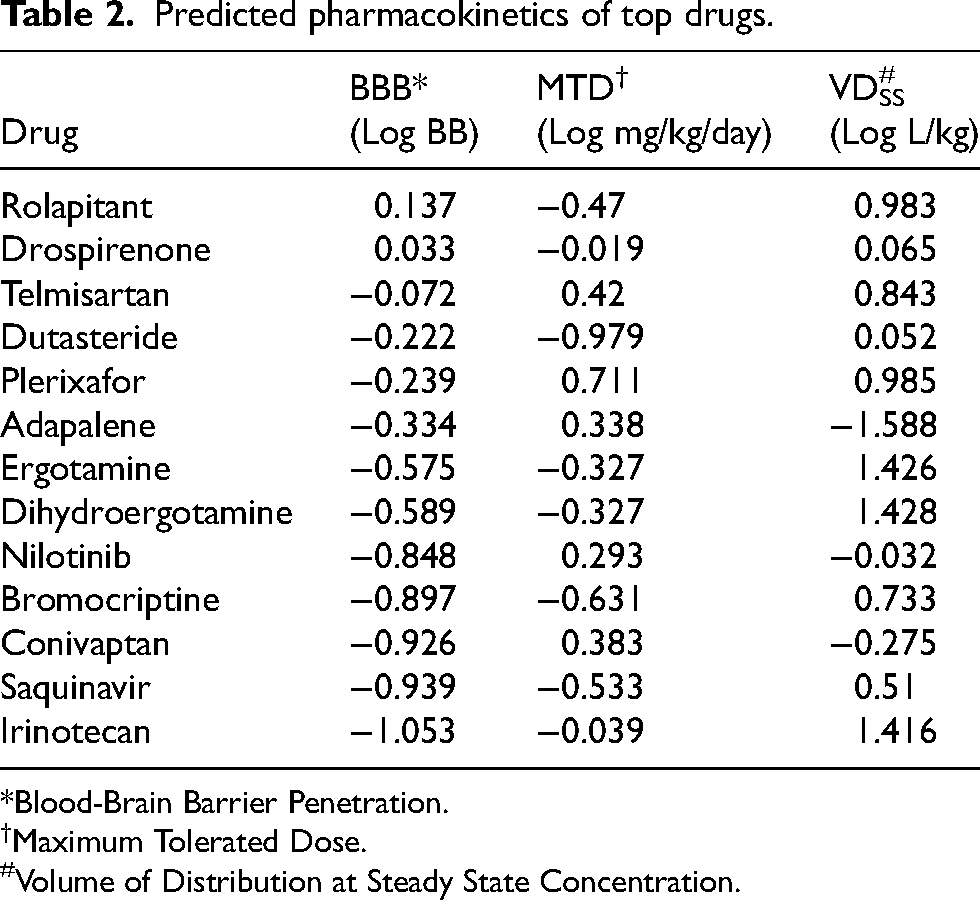
*Blood-Brain Barrier Penetration.
Maximum Tolerated Dose.
Volume of Distribution at Steady State Concentration.
Despite the strong binding of Ergotamine, and Dihydroergotamine to multiple AD targets, their modest brain penetration severely limits their clinical use. Theoretically, ergotamine and dihydroergotamine can cross into the brain of AD, in which BBB integrity is compromised. Yet, even if they cross, Ergotamine and Dihydroergotamine have sustained vasoconstrictor actions, and are contraindicated in patients with coronary, cerebral, and peripheral vascular disease, thus potentially worsening AD symptoms. It is surprising that Ergotamine has been recommended as potential treatment of AD. 26 Like Ergotamine and Dihydroergotamine, Bromocriptine poorly penetrates the BBB, and it is surprising, therefore, that these ergot alkaloids have been suggested as a potential treatment of AD. 27 Likewise, other top candidates could also be counterproductive. Irinotecan is a toxic chemotherapeutic agent, and not recommended for AD patients. Saquinavir is a HIV-1 protease inhibitor that fails to penetrate the BBB. Adapalene is synthetic retinoid used topically in the treatment of acne; however, the oral route presents high toxicity, and could also be unrecommended for AD. As such, top-ranking candidates may not necessarily be effective in the treatment of Alzheimer's disease (AD), highlighting the need for careful evaluation and critical analysis.
Discussion
In this study, we virtually screen a library of approved drugs against 11 AD targets using molecular docking. Then, we analyze the top 10 drugs for each target for recurrence. Top drugs that bind 3 or more AD targets are: (1) Ergotamine, (2) Dihydroergotamine, (3) Dutasteride, (4) Drospirenone, (5) Nilotinib, (6) Adapalene, (7) Conivaptan, (8) Bromocriptine, (9) Rolapitant, (10) Irinotecan, (11) Plerixafor, (12) Telmisartan, and (13) Saquinavir (Figure 2A). These drugs can be classified broadly into ergot alkaloids, steroid hormones, retinoids, NK1 receptor antagonists, antivirals, antidiuretic hormone (ADH) inhibitors, and angiotensin receptor blockers (ARB) as discussed henceforth. The ergot alkaloids, and steroid hormones are indiscriminate binders likely driving the disease, and unfit for AD repurposing, as discussed hereafter. The remaining drugs potentially halt disease progression, but Nilotinib, Adapalene, Irinotecan, and Saquinavir suffer from high toxicity or BBB impermeability, and are bad candidates for AD repurposing. Good candidates include ARBs (i.e., Telmisartan and Candesartan), NK1 receptor antagonists (i.e., Rolapitant and Netupitant), and ADH inhibitors (i.e., Conivaptan and Tolvaptan). The good candidates readily cross the BBB and/or protect its integrity, and exhibit low toxicities. Interestingly, the good candidates reduce hypertension, a comorbid trait associated with AD, by blocking angiotensin II response that is also involved in learning and memory.57–60 The good candidates also reduce inflammation and are orally available.
Ergotamine, Dihydroergotamine, and Bromocriptine (Ergot alkaloids)
Among the top 10 ligands, the ergot alkaloids, Ergotamine, Dihydroergotamine, and Bromocriptine, emerge as highly recurrent in both frequency distribution and heatmap analyses. The ergot alkaloids are dopaminergic receptor agonist associated with vasoconstriction, and can exacerbate hypertension associated with AD. 61 In addition, these drugs poorly cross the BBB, which is a critical requirement for treating this central nervous system (CNS) disease. 62 While the CNS effect of Ergotamine, and Dihydroergotamine in AD patients is limited due to poor brain penetration, it is expected to produce vasoconstriction with adverse effects such as ischemic hypoxia (i.e., ergotic ‘brain gangrene’) in AD patients with compromised BBB. 63 Overall, the use of ergot-alkaloids, such as Hydergine, an Ergoloid Mesylate, have shown no long-term benefit in the treatment of dementia.64,65 On the opposite, epidemiological studies link AD with migraine, and the use of ergot alkaloids. 66 As such, iatrogenic AD could be linked with repeated administration of ergot alkaloid in migraine patients suffering from compromised BBB. AD has also been linked to fungal infections with Aspergillus spp., that produce ergot mycotoxins.67,68 As such, sporadic AD can be linked with prolonged exposure to ergot-alkaloids found in airborne fungal spores, that cross compromised BBB or bypass it entirely in the nasal cavity. 69 To add insult to injury, Ergot alkaloids also exhibit anti-prolactin effects, and suppress proliferation of oligodendrocyte precursor cells in the CNS. 70 These findings all question the recent proposals to repurpose ergot alkaloids as a potential drug against AD.26,27 Notably, the clinical trials using multiple ergoloid mesylates,64,65 do not support assertions that dihydroergocristine mesylate71,72 and Bromocriptine mesylate 72 attenuate AD pathologies in mice. As such, the ergot alkaloids, which together potentially bind all 11 AD protein targets (i.e., Aβ, AChE, APOE4, AβPP, BACE, BChE, BDNF, MAPT, PSEN1, PSEN2, and SNCA), do not halt AD progression, but rather drive it.
Dutasteride and drosperinone (steroids)
The binding sites can also accommodate steroid hormones, and the top 10 ligands include Dutasteride and Drospirenone with high recurrence. Dutasteride is a 5α-reductase inhibitor, and Drospirenone is a synthetic progesterone. These sterols readily cross the BBB; however, clinical findings associate them with an increased risk of AD. Notably, men taking dutasteride, or finasteride, were at increased risk of dementia (HR 1.10) in general, and of AD (HR 1.28) in particular. 73 Persistently, AD has also been associated with BPH (HR 1.10–1.20), and 5α-reductase inhibitor users, compared with the general population 74 As such, iatrogenic AD could be linked to prolonged administration of dutasteride in men diagnosed with BPH. 73 Drospirenone is a synthetic progesterone used in combined hormonal contraception, and in postmenopausal hormone replacement therapy. Both estrogen (Figure 1C) and progesterone (Figure 1B-D, F, J-K) independently and interactively modulate AD neuropathology. 75 When co-administered, progesterone blocks the beneficial effect of estrogen on Aβ accumulation in ovariectomized mice. Also, co-administration of Drospirenone and estrogen fail at improving working memory of ovariectomized rats 76 AD has also been linked with postmenopausal hormone therapy, both with estrogen (HR 1.05–1.14) and with estrogen-progestogen combinations (HR 1.13–1.21). 77 As such, iatrogenic AD could be linked to administration of (combined) hormone replacement therapies in postmenopausal women. Finally, Drospirenone-containing pills are contraindicated in patients susceptible to venous thromboembolism, due to a high risk of blood-clots in the arms or legs, lung, heart, and brain, compared to other progestin-containing pills. 78 As such, steroid hormones which potentially bind 6–7 out of the 11 AD protein targets (Dutasteride: Aβ, BDNF, PSEN1, PSEN2, SNCA; and Drospirenone: AChE, BChE APOE4, AβPP, PSEN2, SNCA; Estradiol: APOE4), do not halt AD progression, but rather drive it.
Nilotinib
Nilotinib is a kinase inhibitor used for the treatment of chronic myeloid leukemia with Philadelphia chromosome. 79 It penetrates the BBB, and several studies highlight the potential of Nilotinib in the treatment of AD. Notably, Nilotinib has been shown to reduce Aβ burden, 80 hippocampal volume loss, 81 and inflammatory markers. 82 Moreover, Nilotinib has demonstrated the ability to restore synaptic dysfunction in human embryonic stem cells. 83 Finally, one less favorable study found Nilotinib to significantly increase BACE1 protein expression, without affecting that of AβPP. 84 Nilotinib has been registered for phase III clinical trial by KeifeRx in December 2021. Yet, to the best of our knowledge, the trial has not started, and nilotinib for AD treatment no longer appears in the company pipeline. 85 Nilotinib safety in AD patients has been questioned, with major concerns expressed about ca,rdiovascular side effects (i.e., arrhythmias, hypertension, and heart failure), which can compound AD comorbidities. 86 As such, Nilotinib potentially binds 6 of the 11 AD protein targets (i.e., Aβ, AβPP, BDNF, MAPT, PSEN2, and SNCA), partially halting disease progression; however, major adverse effects limit its use.
Adapalene (retinoids)
Adapalene is a third-generation topical retinoid approved for the treatment of acne and cervical neoplasia. Like other retinoids, adapalene penetrates the BBB, which have shown promising potential in AD treatment. 87 Notably, Adapalene completely inhibits Aβ aggregation. 88 Adapalene's neuroprotective effects against oxidative stress in vitro remain debated, with one study supporting its efficacy 89 and another refuting it. 90 However, Adapalene topical administration has been linked with intracranial hypertension, 91 and oral administration is contraindicated due to safety concerns. 92 As such, Adapalene potentially binds 5 of the 11 AD protein targets, Aβ, AChE, AβPP, BDNF, and SNCA, indicating its potential to partially modulate disease progression. However, oral toxicity critically limits Adapalene repurposing for AD treatment.
Irinotecan
Irinotecan is a topoisomerase inhibitor, used in the treatment of cancers, like metastatic colorectal carcinoma and pancreatic adenocarcinoma. Irinotecan crosses BBB readily but is significantly toxic. The drug's mechanism of action involves DNA damage and cell death, which is beneficial for cancer treatment, but harmful for neurodegenerative diseases like AD. 93 Despite potential binding to AβPP, PSEN2, and SNCA, Irinotecan is not a candidate for AD repurposing.
Saquinavir, Indinavir, and Daclastavir (Antivirals)
Saquinavir and Indinavir are a HIV protease inhibitor used for the treatment of HIV infection. Likewise, Daclastavir antiviral drug that targets the non-structural protein 5A (NS5A) used in the treatment of hepatitis C virus infection. These drugs are ineffective at treating viral infection in the brain because they do not cross the BBB. 94 Saquinavir, potentially targeting Aβ, BACE, BChE, PSEN1; Indinavir targeting PSEN1; and Daclastavir targeting BDNF, are not candidates for drug repurposing in AD, unless administered intranasally to bypass the BBB.
Plerixafor
Plerixafor is a CXCR4 antagonist used in combination with granulocyte-colony stimulating factor to mobilize hematopoietic stem cells to the peripheral blood. Preclinical studies have shown that plerixafor can cross the BBB, 95 and antagonize CXCR4 expression which is significantly increased in AD patients. 96 Moreover, subcutaneous administration of Plerixafor in a mouse model of AD improved cognition, and reduced neuroinflammation. 97 As such, Plerixafor potentially binds Aβ, MAPT, and AβPP, and it is a candidate for repurposing in AD. 98 Notably, Plerixafor is orally unavailable, which could limit its use.
Conivaptan and Tolvaptan (antidiuretic hormone inhibitor)
Conivaptan and Tolvaptan are ADH inhibitors, used to raise serum sodium levels in euvolemic and hypervolemic hyponatremic patients. Conivaptan exhibits unfavorable BBB penetration, but exerts neuroprotective effects against oxidative stress mechanisms in AD. 90 Conivaptan potentially reduces aggregation in model systems by inhibiting Hexokinase-1 and 14–3-3G/γ protein interaction. 99 Notably, conivaptan attenuates cerebral edema and BBB disruption following cardiac arrest, 100 and it reduces blood cytokine levels in rat models of autoimmune encephalomyelitis. 101 Tolvaptan also improves psychomotor speed domain, and is less studied than Conivaptan. 102 Conivaptan is not without it risks either, of headache, and infusion site reactions, and too rapid correction of hyponatremia can cause fatal osmotic demyelination syndrome. 103 Finally, Conivaptan is readily available orally. 104 As such, both ADH inhibitors: (1) Conivaptan which potentially binds to Aβ, BChE, BDNF, MAPT, and PSEN2, and (2) Tolvaptan which potentially binds to BDNF and PSEN2, are good candidates for drug repurposing in AD.
Rolapitant and Netupitant (NK1 receptor antagonists)
Rolapitant and Netupitant are selective neurokinin 1 (NK1) receptor antagonist, used in the treatment of chemotherapy-induced nausea and vomiting. Rolapitant readily crosses the BBB, and blocks the action of tachykinin peptides, such as substance P which plays an important role in neuroinflammation. 105 Rolapitant is also approved for treating pulmonary arterial hypertension, by improving blood flow and reducing hypoxemia. While the oral formulation of Rolapitant (i.e., Varubi) was withdrawn in 2018 due to reports of anaphylactic shock, Netupitant remains orally available. Rolapitant potentially binds to BDNF, BACE, and AChE, and Netupitant to PSEN2 and BChE, and as such NK1 receptor antagonists are good candidates for drug repurposing in AD. 106
Telmisartan, Candesartan, Azilsartan (Angiotensin receptor blockers)
Telmisartan and Candesartan are angiotensin II receptor blockers (ARB), used orally in the treatment of hypertension, diabetic nephropathy, and heart failure. Remarkably, ARB use is associated with a reduced incidence of cognitive impairment, dementia, and AD in white men and women. 107 Likewise, Telmisartan is associated with a lower risk of AD in African Americans, 108 and with a lower risk of dementia in East Asians suffering from diabetes and hypertension. 109 Notably, use of ARB, and to a lesser degree use of β-adrenoceptor blockers, 107 but not use of angiotensin converting enzyme (ACE) inhibitors, 110 reduce AD incidence. In AD mice models, Telmisartan also alleviates AD-related neuropathologies and cognitive impairments, 111 and intranasal Telmisartan significantly reduced amyloid burden in the cortex and hippocampus. 112 While all ARBs penetrate the BBB, the highly lipophilic Telmisartan attains brain concentration 10-fold higher than those of Losartan and Irbesartan, 113 and Candesartan restores BBB dysfunction. 114 Several studies have highlighted disproportionate neuroprotective benefits of Telmisartan and Candesartan over other ARBs without understanding their mechanism. Here, we propose potential mechanisms through binding multiple protein targets associated with AD. Other potential mechanisms include high lipophilicity and BBB restoration through APOE4-binding. 115 Telmisartan potentially targets APOE4, BChE, and MAPT, Candesartan targets BACE, and Azilsartan targets AChE, and as such these ARB are good candidates for drug repurposing in AD.
Limitations
As a potential limitation to this study, it is crucial to note that our findings are based on computational work. Therefore, the beneficial or deleterious effect of these drugs in AD treatment remains to be validated experimentally and clinically. Experimental validation is required to evaluate the binding affinity of top drugs to protein targets of AD. In addition, clinical validation is required to evaluate the overall effect of good candidates for repurposing.
As another potential limitation to this study, multitarget drugs do not necessarily indicate a better chance at AD repurposing. In fact, multitarget drugs are indicators of less discriminate selectivity, and of more potential toxicity. This could help explain why highly multitarget drugs, like ergot alkaloids, and steroids are not good candidates for AD repurposing. Conversely, drugs targeting only one AD protein could be too selective and restrain their repurposing potential. Thus, a balance between too much selectivity, and too little selectivity is required to identify good candidates. In fact, ergotamine has been proposed to cure everything from colon cancer, 116 to arbovirus infection, 117 and in-silico it binds to 24 proteins of the coronavirus, 118 including coronavirus protease, 119 and coronavirus helicase, 120 as well as human MCL-1, 16 NRP1, 121 sortilin, 122 Histamine N-Methyl Transferase, 123 TGF-β receptor type 1 (ALK5), 124 casting immense doubt on the specificity of ergot alkaloids. The latter paper calls out the false positives. We believe ergot alkaloids are not specific enough for repurposing in AD.
Finally, as a limitation of this paper, the good candidates for drug repurposing in AD include the top drugs, but not automatically any other members of their drug families.
Conclusion
Good candidates for drug-repurposing in AD include: (1) The antidiuretic hormone inhibitor, Conivaptan and Tolvaptan; (2) The NK1 receptor antagonist, Rolapitant, and Netupitant; and (3) The angiotensin receptor blocker, Telmisartan, and Candesartan. Their selective multitarget potential, antihypertensive and anti-inflammatory properties, BBB penetration or protection, and oral availability underscores their suitability for further investigation.
Supplemental Material
sj-docx-1-alz-10.1177_13872877251387167 - Supplemental material for Virtual screening of drugs against multiple targets of Alzheimer's disease
Supplemental material, sj-docx-1-alz-10.1177_13872877251387167 for Virtual screening of drugs against multiple targets of Alzheimer's disease by Karin Ben Zaken, Samuel Mesfin, Naamah Bloch and Abraham O Samson in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
We thank the Katz foundation for a generous grant to AOS.
Author contribution(s)
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant to AOS from the Katz family foundation, and the Ginsburg Family foundation.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Supplemental figures and tables are available on the journal website.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.