
Abstract
Alzheimer's disease is a major cause of dementia, that affects approximately 7–8% of people aged 65 years and older (according to WHO & Alzheimer's Disease International) and thus is a major concern in public health world-wide. This review chronicles the foundational research and translational trajectory leading to the development of donanemab, a monoclonal antibody targeting pyroglutamyl amyloid-β (Aβ3pE-X) peptides, recently approved for the treatment of early Alzheimer's disease (AD). We trace a 30-year arc from the biochemical identification of Aβ species to the recognition of Aβ3pE-42 as a predominant pathological isoform in AD and Down syndrome brains—a fact still underrecognized among clinicians and researchers. We highlight key breakthroughs in antibody generation, Aβ peptide biochemistry, and resistance to enzymatic degradation. Mechanistic distinctions between donanemab (Kisunla®), and lecanemab (Leqembi®) are also explored, along with therapeutic implications for targeting specific Aβ species at preclinical stages of disease. The review emphasizes how persistent biochemical research, fueled by intellectual curiosity, serendipity, and rigorous experimentation, has culminated in clinical proof-of-concept for the amyloid hypothesis. In addition to its molecular specificity, donanemab's development underscores a critical shift toward precision medicine in Alzheimer's care. Its clinical validation, though limited in scope, reinforces the need for scalable and affordable interventions that can address the growing global burden of dementia. We expect these therapeutic antibodies to contribute to reducing the global burden by ceasing the disease progression in a preclinical stage now that new methods for fluid biomarkers are becoming available. As the global population ages, understanding and addressing AD has become a top priority in neuroscience and public health.
Keywords
The development trajectory of donanemab—one of the two disease-modifying monoclonal antibodies now employed in the treatment of early Alzheimer's disease (AD)1,2—can be traced back to 1992, when Selkoe and colleagues identified amyloid-β (Aβ) 1-40 as the predominant Aβ species isolated from AD brains. 3 Utilizing advanced two-dimensional reversed-phase high-performance liquid chromatography (HPLC) coupled with mass spectrometry, their study was broadly recognized as a confirmation of earlier findings by Glenner et al., who initially reported the N-terminal sequences of vascular and parenchymal Aβ peptides from human brain4–6 Notably, Selkoe's team also detected a minor presence of Aβ3pE-40 (where pE denotes pyroglutamate), a finding that would later prove to be of major importance.
Driven by scientific intuition, Saido, the late Ihara and colleagues hypothesized that Aβ3pE-X species might be critically involved in AD pathogenesis. This was grounded in the notion that converting Aβ1-X to Aβ3pE-X results in the loss of one positive and two negative charges at the peptide's N-terminus, potentially altering its biochemical behavior (Figure 1)7–9 To explore this, our team applied a method we had previously developed to generate antibodies against specific protein termini using 5- to 6-mer synthetic peptides (for example, pEFRHD for Aβ3pE-X, and DAEFR for Aβ1-X) conjugated via cysteine to keyhole limpet hemocyanin.10–15 First, using this approach, we created an antibody selective for Aβ3pE-X and analyzed aged human brain tissue using immunohistochemistry and Western blotting. 7 We found that Aβ3pE-X, but not Aβ1-X, was the dominant species in the cerebral cortex, including in the frontal, temporal, and occipital lobes. The latter was rather quantitatively less significant.
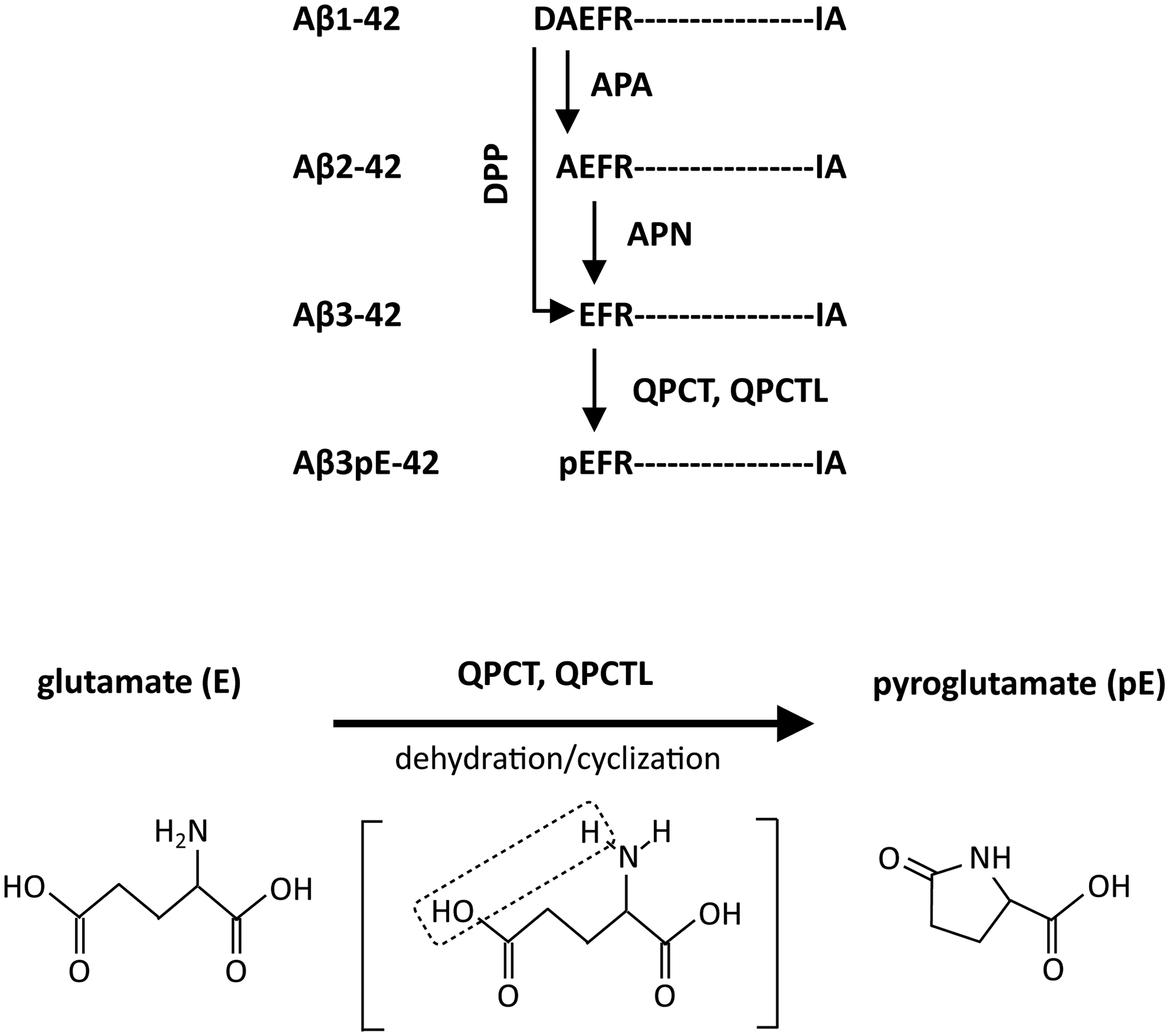
Possible pathway(s) for generation of Aβ3pE-42 from aβ1-42. DPP: Dipeptidyl peptidase 4; APA: Aminopeptidase A (acidic amino acid); APN: Aminopeptidase N (neutral amino acid); QPCT: glutaminyl-peptide cyclotransferase or glutamyl cyclase; QPCTL: glutaminyl-peptide cyclotransferase-like or glutamyl cyclase-like enzyme. The N3E is the only final precursor for N3pE. The lower panel shows that the conversion of E to pE results in the loss of one positive and one negative charges. Reproduced with minor modifications from Iwata et al. (CC BY 4.0). 17 .
Subsequently, we developed antibodies capable of distinguishing between various N- and C-terminal Aβ structures each other, confirming that Aβ3pE-42 is the most abundant form in autopsied brains from AD patients, followed by Aβ1(iso-D)-42 and Aβ1(D-Asp)-42 (Figure 2)8,16,17 The amount of Aβ3pE-42 correlated better with phospho-tau load in AD and age-matched control brains rather than that of the others. 18 Importantly, phospho-tau pathology correlates well with cognitive impairment as measured by NIA-AA or DSM-5 definitions.19–22 Deposition of Aβ3pE-42 thus appears to be associated with AD pathogenesis. These species exhibit a shared biochemical trait: resistance to major mammalian aminopeptidases. Our findings, along with those of others, confirmed the predominance of Aβ3pE-42 in both AD and Down syndrome (DS) brains.8,9,17,23–26 The resistance of plaque-associated peptides isolated from AD brains in the earlier days to Edman degradation 27 further supports their presence as major pathological forms.
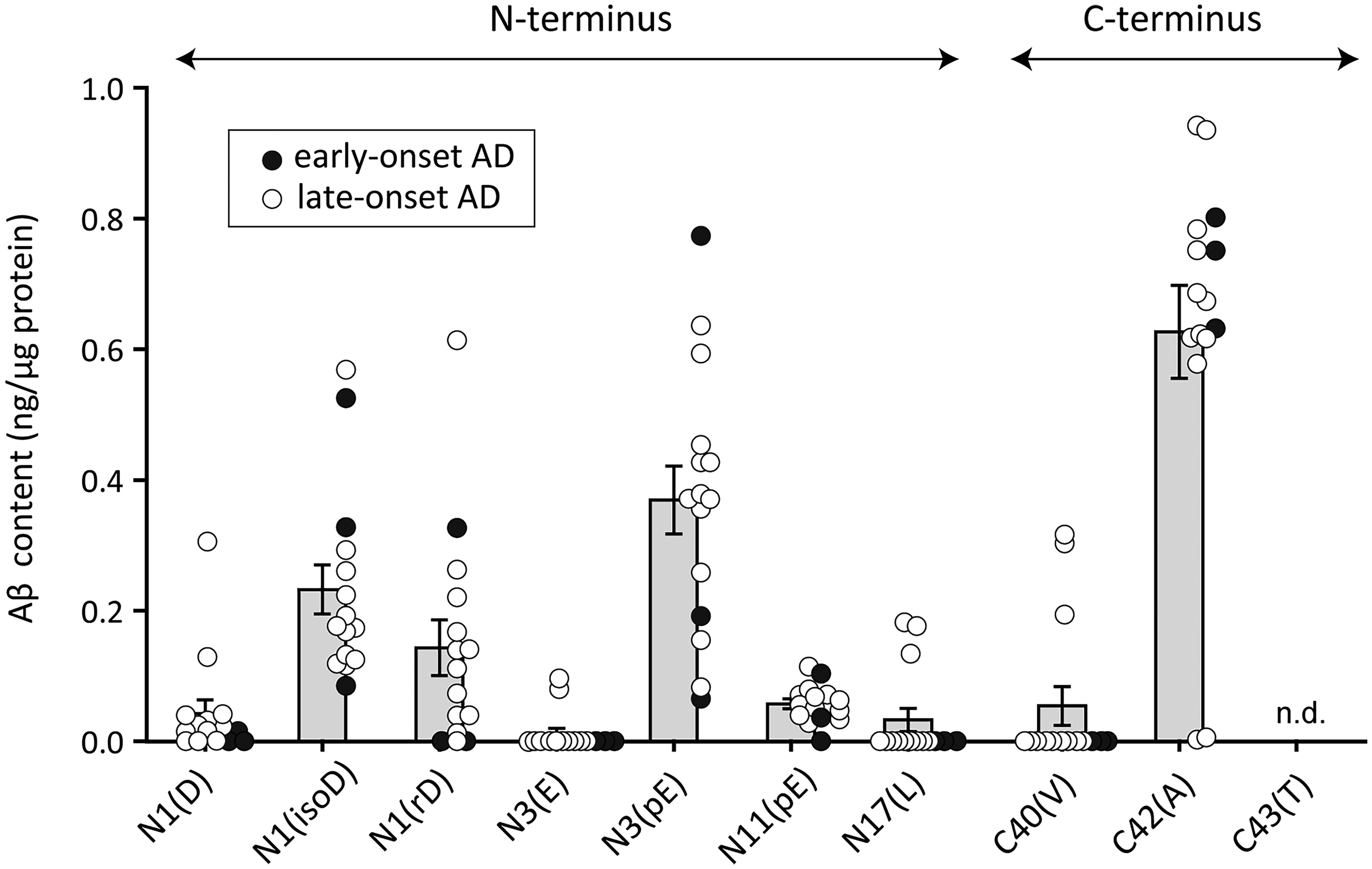
Quantified terminal structures of Aβ extracted from AD brain. Western blot analyses of various Aβ species were performed and quantified using different amounts of corresponding synthetic Aβ peptides: Aβ1(D)-42, Aβ1(isoD)-42, Aβ1(rD)-42, Aβ3(E)-42, Aβ3(pE)-42, Aβ11(pE)-42, Aβ17(L)-42, Aβ1(D)-40, Aβ1(D)-42, and Aβ1(D)-43. isoD, isoaspartate; rD, rectus (D-form) aspartate; pE, pyroglutamate. These experiments were performed as previously described.7,16,17 Samples were obtained from early-onset AD patients (age at death: 45 ± 1.0 years) and late-onset AD patients (74 ± 6.6 years). Human brain tissues were kindly provided by Dr. David M. A. Mann (University of Manchester). Reproduced with minor modifications from Iwata et al. (CC BY 4.0). 17 .
Why did the 1992 paper 3 overlook Aβ3pE-42 or even Aβ1-42 isoforms in their biochemical analysis? The group led by Younkin gave a partial answer: It was necessary to heat the HPLC column up to 50°C to elute Aβ1-42. 28 We then found that use of a basic buffer, triethylamine, in combination with a low-molecular-weight detergent, betaine, enables us to fully isolate Aβ3pE-42 in the process of synthesizing and purifying various forms of the internally radiolabeled AβX-42 peptide (Table 1).8,17,29 Satoshi Tsubuki, specialist of analytical and synthetic organic chemistry, Laboratory for Proteolytic Neuroscience, RIKEN CBS, played a major role in determining these conditions. Moreover, we were lucky to have used a high concentration of formic acid to extract Aβ from human brain tissue as this is the most stringent way to isolate Aβ from human brain amyloids.4–6 If we had not realized such appropriate analytical techniques, we would not have been able to detect Aβ3pE-42 from human brain tissues constantly. Other groups have also showed observations consistently similar to ours.23–26,30
Recovery of various Aβ variants for elution from reversed phase HPLC under different conditions.

TFA: Trifluoroacetic acid; TEA: Triethyl amine.
Reproduced with minor modifications from Iwata et al. (CC BY 4.0). 17
Interestingly, use of guanidine hydrochloride, not formic acid, was sufficient to fully extract Aβ from the brains of mouse models of Aβ amyloidosis (such as AppNL −F , AppNL −G−F and APP23),31,32 and the major isoforms therein was Aβ1-40/42.17,23,31 Thus, the physicochemical properties of pathological Aβ differ between AD and mouse model brains. This difference likely suggests that conversion of Aβ1-42 to Aβ3pE-42 takes a long time, probably years to decades, in human brain tissue as Aβ deposition is known to precede the disease on-set by more than two decades. 33 Consistently, pathological analyses of DS brains demonstrated presence of diffuse plaques and absence of neuritic plaques in the early childhood (before 10 years), 34 and presence of Aβ1-X and Aβ3pE-X in small subsets of compacted, neuritic plaques beginning around age 30 and rose with age, the latter isoforms always exceeding the former. 26 Given the short lifespan of mice (typically under three years), it remains challenging to investigate the long-term biochemical conversion of Aβ1-42 to Aβ3pE-42 in vivo, by which Aβ1-42 is converted to Aβ3pE-42. In fact, no mouse models developed thus far accumulate Aβ3pE-42 as a major Aβ isoform to our knowledge. Since glutamine (Q) is much easier to convert to pE than glutamate (E), 35 we made an Aβ protein precursor (APP) cDNA construct, in which the first two amino acids of Aβ part are deleted and the third E was converted to Q, and expressed APP Δ(DA)−Q−F in primary neurons, leading to generation of Aβ3pE-40/42. 36 However, App knock-in mice carrying these mutations together with the Beyreuther/Iberian mutation, AppΔ(DA)−Q−F, failed to accumulate Aβ3pE-42 17 presumably due to the following reasons.
In general, the selective deposition of Aβ3pE-42 in human brain is attributed to their high oligomerization/fibrillization/aggregation-prone properties as examined by in vitro paradigms.37–42 However, this may not be the only reason because Aβ11pE-42 and Aβ17-42 (p3 fragment), more hydrophobic and aggregation-prone than Aβ1-42 and Aβ3pE-42,43–52 are relatively minor isoforms in aged human and AD brains (Figure 2).8,16,17,53 We thus examined the in vivo half-lives of various AβX-42 isoforms in rat hippocampus using internally radiolabeled Aβs and found that most Aβ variants lacking the N-terminal amino acid(s) except Aβ3pE-42 were more short-lived (half-life: approximately 10 min) than Aβ1-42 (half-life: approximately 18 min). In contrast, the half-life of Aβ3pE-42 was as long as 90 min (Table 2). 17 These observations likely explain why Aβ3Q-42 was quickly proteolyzed before conversion to Aβ3pE-42 in vivo and why Aβ3pE-42 dominantly accumulate in aged and AD brains. It is possible that Aβ3pE-42 aggregated or became associated with some pathological structures such as dystrophic neurites 54 or a complement complex, C1q,55,56 faster than the other isoforms. The reason for Aβ11pE-42 and Aβ17-42 isoforms being relatively minor isoforms in AD brains remains elusive, but we speculate that autophagy followed by endocytosis57,58 might play a role in their clearance although we have no experimental evidence.
In vivo half-lives of various Aβ species.
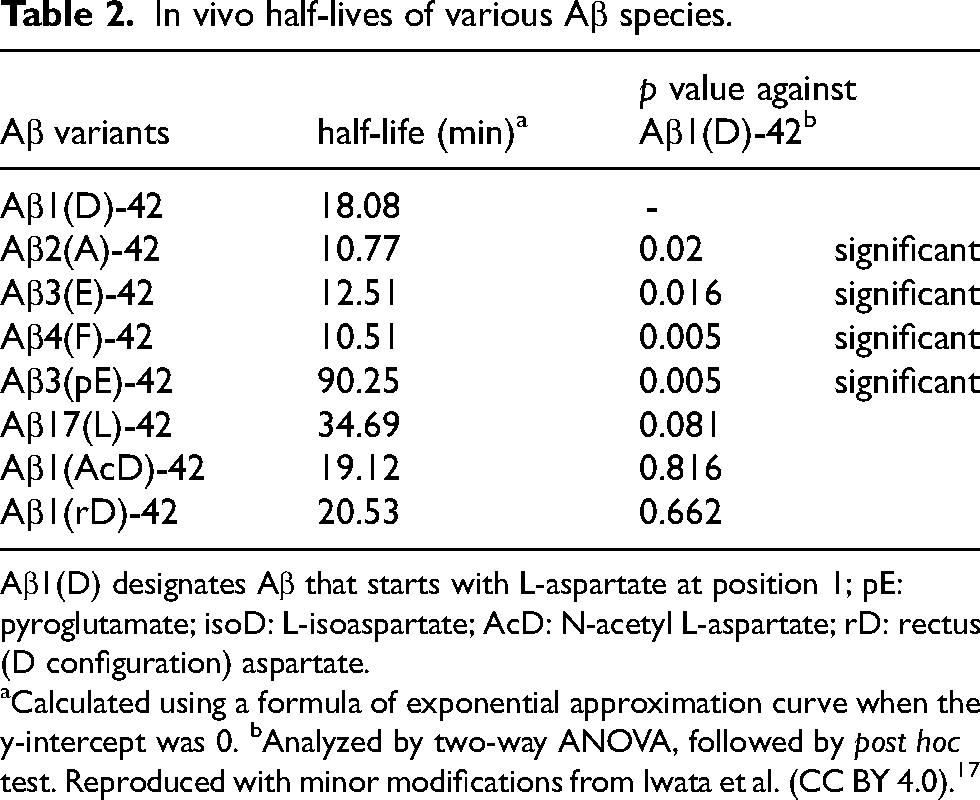
Aβ1(D) designates Aβ that starts with L-aspartate at position 1; pE: pyroglutamate; isoD: L-isoaspartate; AcD: N-acetyl L-aspartate; rD: rectus (D configuration) aspartate.
Calculated using a formula of exponential approximation curve when the y-intercept was 0. bAnalyzed by two-way ANOVA, followed by post hoc test. Reproduced with minor modifications from Iwata et al. (CC BY 4.0). 17
It is notable that deficiency of neprilysin, a major Aβ-degrading enzyme mainly presynaptically localized in brain,59–61 resulted in a significant increase of Aβ3pE-40/42 particularly associated with plaque cores in the APP-transgenic mice, APP23, and App knock-in mice, AppNL −F, as examined by immunohistochemistry, Western blotting, and LC-MS/MS following proteolytic digestion. 17 Presumably, neprilysin-independent pathways are, at least in part, involved in N-terminal truncation of Aβ and formation of Aβ3pE-42. Consistently, neprilysin-deficiency increased expression of aminopeptidase N, dipeptidyl peptidase, and glutamyl cyclase-like enzyme in the APP-transgenic mice with aging, 17 all of which may contribute to Aβ3pE-X formation (Figure 1).62–70 It is also important that a number of studies consistently demonstrated an aging-dependent or AD-associated decline of neprilysin expression in the human and mouse brains,59,60,71–76 and that a GWAS-identified mutation in the neprilysin gene, MME, associated with an AD risk 77 perturbs neprilysin-dependent extracellular Aβ degradation (Figure 3). 78
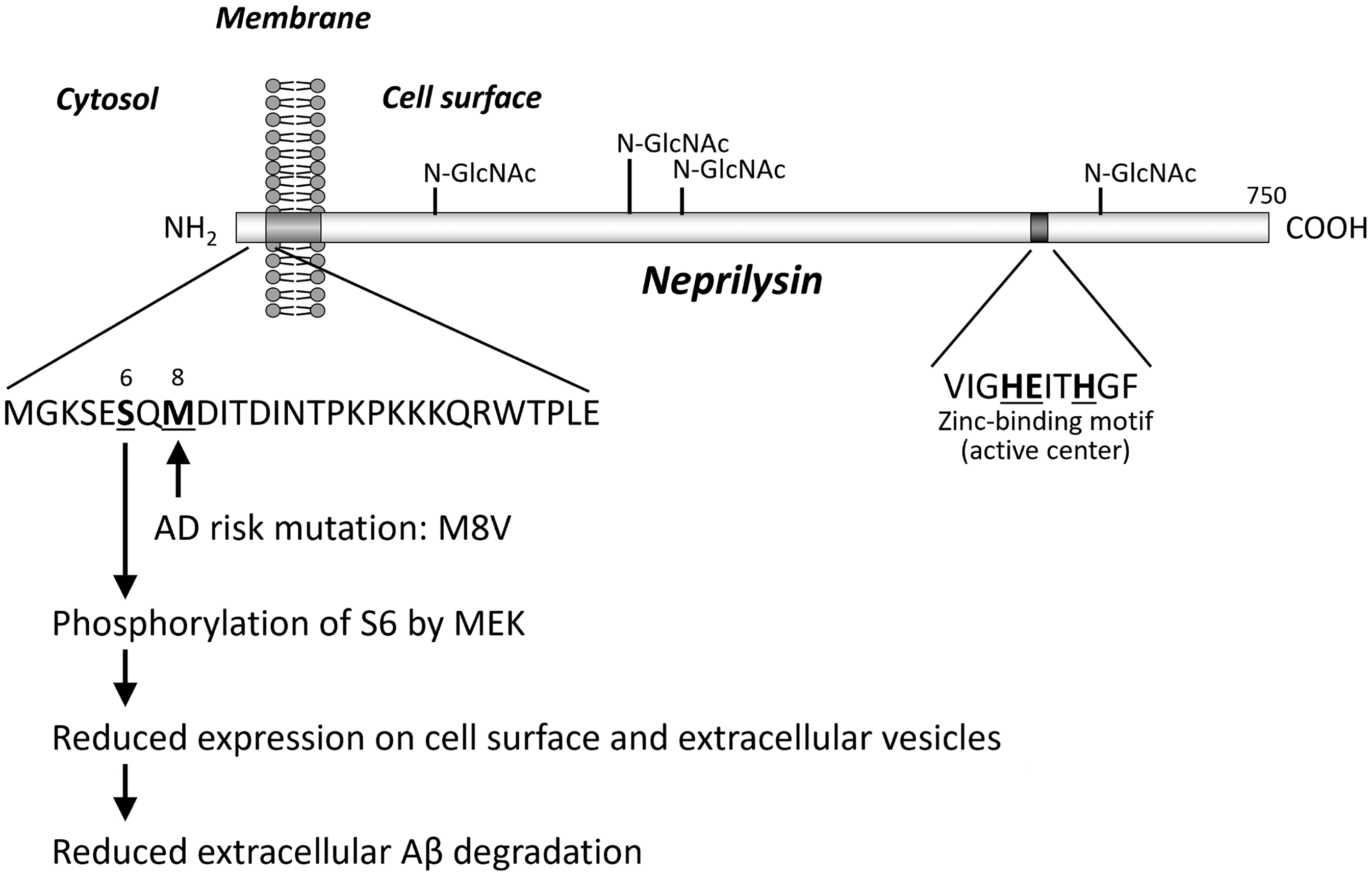
Effect of M8 V mutation on the action of Aβ-degrading enzyme, neprilysin, encoded by MME gene. The M8 V mutation is associated with an AD risk. 77 The mutation reduces extracellular Aβ degradation by suppressing neprilysin expression on cell surface and extracellular vesicles via phosphorylation of S6. 97 MEK: mitogen-activated protein kinase/extracellular signal-regulated kinase. 105 .
In the meantime, Demattos and colleagues, Lilly, USA, created a mouse monoclonal antibody specific to Aβ3pE-X and showed that administration of this antibody removes Aβ plaques from APP-transgenic mouse brains 79 The humanized version of this antibody is donanemab.1,2,17 In the TRAILBLAZER-ALZ 2 trial,2 donanemab demonstrated a modest slowing (35%) of cognitive and functional decline in early symptomatic AD patients, with amyloid-related imaging abnormalities (ARIA) occurring in approximately 24% of treated individuals (Table 3). (A modified titration protocol (delaying the third 700 mg vial) reduced ARIA-E rates to ∼14%.) These results have led to FDA approval under accelerated pathways, reinforcing the feasibility of amyloid-targeting strategies in clinical settings. Because we observed the Aβ3pE-positive vascular amyloid in human brain tissue,7,80 its major side-effect, i.e., ARIA,81–84 was predictable. In any case, the success of lecanemab and donanemab in clinical trials (although their effect was limited)1,2,85,86 provided the first clinically evidenced proof-of-concept for the Aβ hypothesis that had been based on pathological, genetic and experimental observations.87,88 We believe the reason for these immunotherapies being limited in the treatment of early AD is that it is too late to do so when irreversible neurodegeneration has already taken place 33 as neuronal circuits composed of postmitotic cells (neurons) cannot in principal regenerate after degeneration. 8 Thus, we must target the anti-Aβ treatment to preclinical AD rather than the later disease stages.8,89 This would reduce the total number of mild cognitive impairment (MCI) and AD patients and contribute to diminishing the socioeconomical burdens all over the world although the currently approved therapeutics are too expensive to cover the preclinical AD patients that outnumber the MCI and AD patients.90–92 This is why we need to develop orally administerable synthetic low-molecular-weight medications that selectively promote neprilysin-dependent Aβ degradation via activation of somatostatin or dopamine receptors93–97 or inhibit aminopeptidases and glutamyl cyclases. 17 We also expect identification of plasma biomarkers such as phospho-tau would accelerate treatment of preclinical AD.
Comparison of three therapeutic anti-Aβ antibodies.
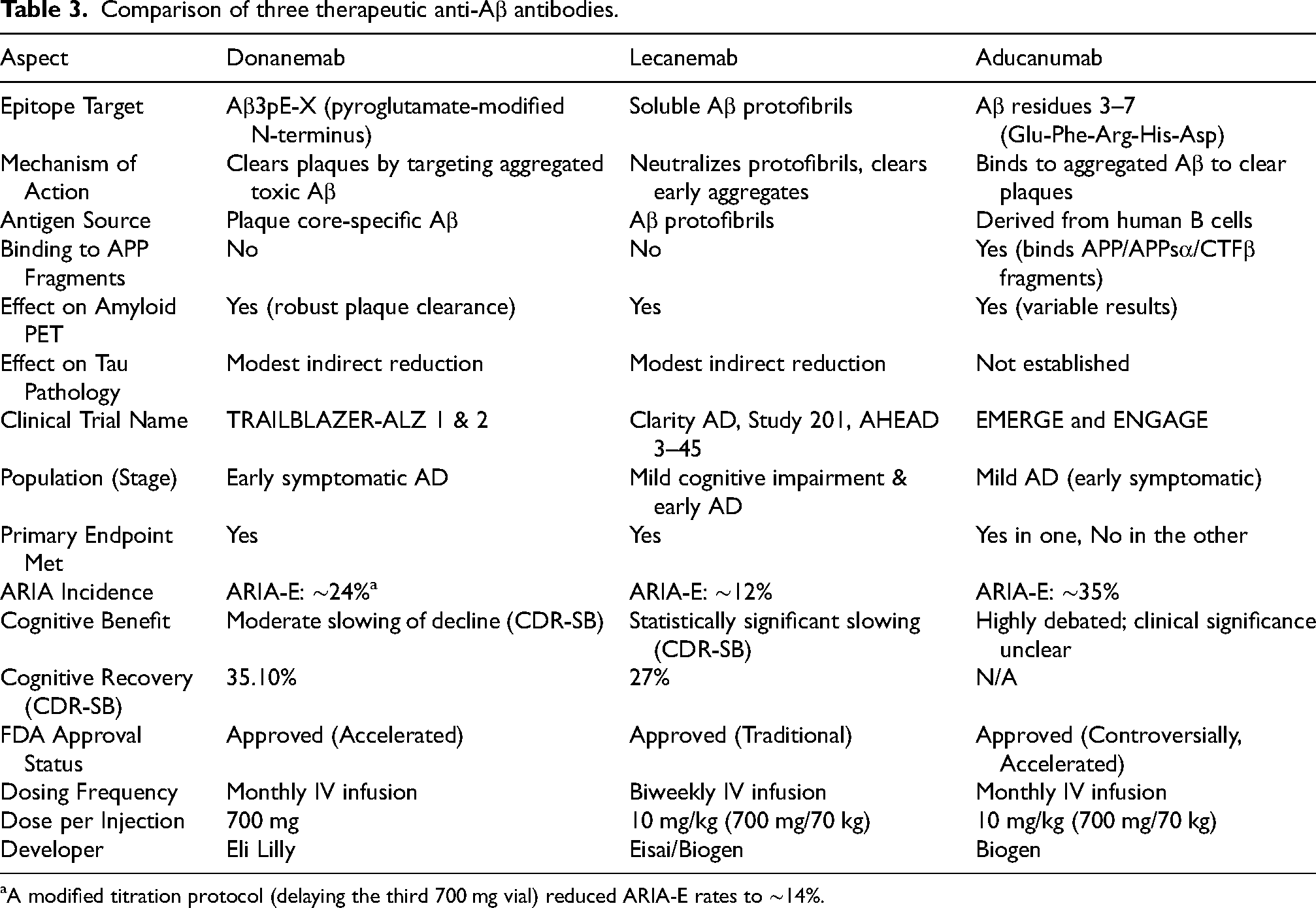
A modified titration protocol (delaying the third 700 mg vial) reduced ARIA-E rates to ∼14%.
Incidentally, lecanemab differs from donanemab with soluble Aβ protofibrils being the original antigen (Table 3).85,86,98–100 However, treatment of early AD patients reduces Aβ amyloidosis as examined by amyloid positron emission tomography (PET) accompanying ARIA in some cases.85,86,101 This implies that the antibody also binds to parenchymal and vascular Aβ deposits in vivo. although we tried to immunostain human Aβ plaques with lecanemab in vain (unpublished observations). Reduction of plasma glial fibrillary acidic protein (GFAP) after lecanemab treatment 85 also implies suppression of the plaque-astrocyte interaction(s). We would like to point out that biological properties of an interaction between a given protein and anything else can drastically differ between the in vitro and in vivo paradigms.17,29 For instance, neprilysin and insulin-degrading enzyme cleave Aβ efficiently in a test tube, whereas they preferentially degrade insoluble and soluble Aβ, respectively, in the mouse brain. 78 Therefore, the mechanisms of action of lecanemab remains unresolved at least in part, but lecanemab and donanemab share one common feature in a manner different from most other therapeutic antibodies including aducanumab. 102 That is, only lecanemab and donanemab avoid binding to AβPP, C-terminal fragment of AβPP developed by β-secretase (AβPP-CTFβ), soluble N-terminal fragment of APP generated by α-secretase (sAβPPα), or other non-Aβ APP fragments physiologically produced and potentially functional in the brain (Figure 4). 8 For instance, an antibody raised against the N-terminus of AβPP-CTFβ 13 turned out to be one of the best antibodies to immunostain Aβ plaques in the PD-APP transgenic mice. 15 Binding of these antibodies to non-Aβ APP fragments may have caused unexpected side-effect(s).
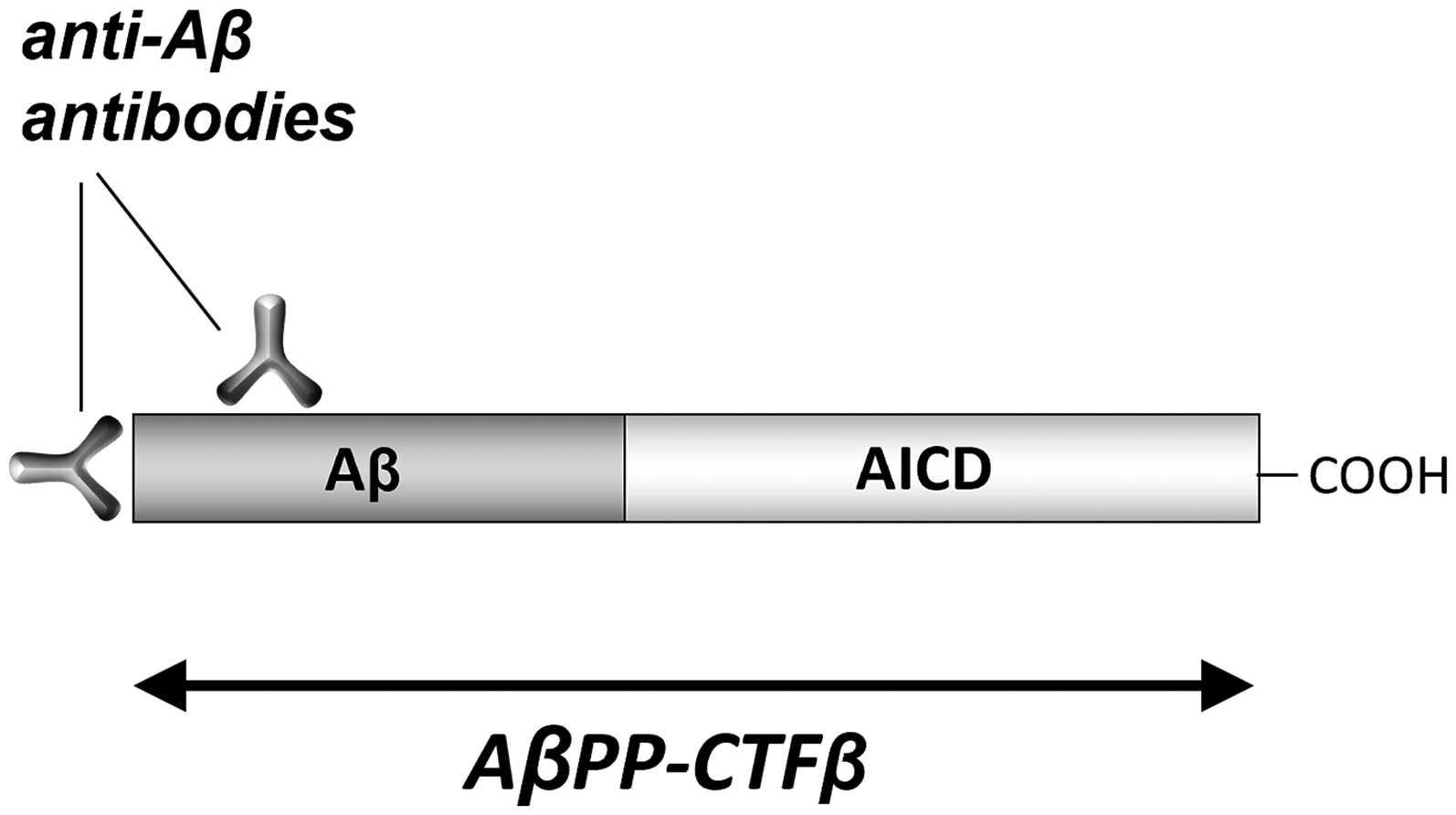
Binding of anti-Aβ antibodies to the CTFβ fragment of AβPP. Modified from Saido TC. 8 Most antibodies raised against Aβ bind to AβPP, CTFβ, sAβPPα, or other non-Aβ, AβPP fragments, but donanemab and lecanemab do not. AICD: AβPP intracellular domain.
We can also predict that combination of lecanemab and donanemab together may result in additive or synergistic effects possibly at lower doses as their epitopes are different, and this could either improve or worsen the side-effects depending on the administration protocols. Generation of mouse models that produce both Aβ3pE-42 and Aβ protofibrils in a sufficient quantity or nonhuman primate models of Aβ amyloidosis30,103,104 that live much longer than mice may help address this issue without experimenting on humans.
In summary, development of donanemab has stemmed from a number of biochemical and immunochemical studies over 30 years since 1992, indicating the importance of basic science based on intellectual curiosity and serendipity. In addition, we would like to point out that the efforts by Ronald Demattos and colleagues to develop monoclonal antibodies to Aβ3pE-X, 79 together with development of lecanemab by Lars Lannfelt and colleagues,98,100 made a major break-through for establishing a clinical proof-of-concept for the Aβ hypothesis in the etiology of AD.87,88 The God has indeed been in the details.
Footnotes
Acknowledgments
This work was supported by research grants from RIKEN, Special Coordination Funds for promoting Science and Technology of STA, CREST, Ministry of Health and Welfare, Ministry of Education, Science and Technology, Chugai Pharmaceutical Co., Mitsubishi Chemical Co., Takeda Chemical Industries, and AMED under Grant Number JP20dm0207001 (Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS)).
During the preparation of this work the authors used ChatGPT4 in order to improve the English writing originally created by the author. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Author contributions
Funding
Funding sources are acknowledged in the Acknowledgements section.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.