
Abstract
An all-encompassing framework seems necessary for understanding brain health in the context of neurodegeneration, particularly due to Alzheimer's disease (AD). We argue that current views, dominated by the amyloid-beta hypothesis, oversimplify the complexity of brain degeneration. We propose a multi-scale, multi-entity approach, treating the brain as a complex system composed of interacting subsystems across various scales, from the nanoscale (genes and proteins) to the macroscale (cognition) and beyond. By redefining brain health through mathematical complexity, we emphasize the importance of integrating diverse biological, environmental, and lifestyle factors to account for the heterogeneity in AD presentations. This framework suggests that failures at different levels of brain function can lead to cascading effects that influence neurodegenerative trajectories. Additionally, it calls for a shift toward personalized treatment approaches that target multiple yet specific pathological mechanisms affecting an individual. We propose computational models as essential tools for simulating and testing these complex interactions. Through this framework, we aim to provide a deeper understanding of neurodegeneration, and advocate for more comprehensive and multi-factorial approaches in both research and clinical practice to advance the treatment of brain health disorders such as AD.
Keywords
Introduction
Alzheimer's disease (AD) is one of the defining challenges of our time. With its risk doubling every five years after age 65, 1 individuals live under its ominous threat while healthcare systems face mounting financial and social burdens as global populations age. Over the past 20 years, the cost of dementia care has surpassed that of cancer. 2 During this period, while deaths from stroke and heart disease have declined, AD-related deaths have increased by 146%. 3
AD is increasingly and solely being defined as the abnormal accumulation of amyloid-β (Aβ) proteins in the brain. This Aβ hypothesis, formulated in 1984, 4 continues to shape basic and clinical research, diagnostic guidelines 5 and frameworks. 6 Yet the central causal role of Aβ is not supported by a wide variety of evidence at multiple levels. For example, the genetic risk signature for conversion to clinical AD appears unrelated to Aβ. 7 Further, Aβ deposition is present at pathological levels in a large proportion of otherwise cognitively healthy seniors8–10 and is poorly corelated with cognitive deficits.11–13 In fact, it is present in nearly every brain from 25 years of age and over, 9 indicative of the ubiquitous nature of amyloid, yet not conceptually related to a disease state. Once deposed, it is not well aligned with the topographical selectivity of tau accumulation, 14 neurodegeneration, or brain energy hypometabolism. 15 Additionally, while recent Aβ antibody treatments (aducanumab, 16 lecanemab, 17 and donanemab 18 ) have been successful at removing Aβ, they result in small cognitive improvements (±25% delay in cognitive decline over 18 months) which are not of the same magnitude as the amyloid lowering. 19 These modest trial results 20 are also accompanied by brain swelling and microbleeds in a significant number of patients, 21 which brings to question what is the physiological role of Aβ in the brain. There is in fact a remarkable dearth of research into the physiological role of amyloid, given its ubiquity.
Unsurprisingly, there are dissenting voices against the centrality of the Aβ hypothesis. 22 As we reviewed recently, 23 other theories have been put forward to explain neurodegeneration with a clinical profile of AD. Authors have suggested that AD is a tauopathy 24 ; a consequence of brain blood barrier dysfunction, including neurovascular unit insults25–28; due to oxidative stress and inflammation, with secondary deposits of Aβ 29 ; the relative evolution of neuroplasticity failure30,31; or metabolic insufficiency (deteriorating brain glucose metabolism), present decades before any clinical symptoms in those at increased risk, 32 to name but a few.
These molecular and cellular theories must be reconciled with the growing body of epidemiological evidence that underscores the impact of modifiable risk factors on dementia. These risk factors—diabetes, smoking, obesity, hypertension, excessive alcohol consumption, physical inactivity, traumatic brain injury, air pollution, depression, hearing impairment, low education, low social contact, and uncorrected vision loss—putatively account for nearly 45% of the incidence of dementia cases globally. 33
Equating AD solely with an amyloidosis therefore seems overly simplistic, as Aβ alone cannot account for the extensive range of dysfunctions observed throughout the brain nor interact with all risk factors. We propose that the centrality of the Aβ cascade hypothesis should be reconsidered. A more global perspective on brain health based on an appreciation of its complex nature seems essential for comprehending the multifaceted causes of neurodegeneration seen in the pattern of clinical presentations surrounding AD. Although this viewpoint is not entirely new, from the entry of Ball et al. in 1982, 34 through various works across decades35–40 up to the present,41,42 it warrants renewed focus.
As was recently argued by Korczyn and Grinberg, 43 we posit that reconceptualizing AD as a syndrome comprising various subtypes with diverse etiologies and characteristics can significantly advance our understanding. Further possessing a complex framework able to reflect the heterogeneity of its neurodegenerative processes would propel us forward even more. This paradigm shift would underscore the necessity for individualized treatments based on the specific pathological mechanisms present in each patient, with profound implications for the design and conduct of clinical trials.
A complex definition of brain health
Mathematical complexity
Most studies of AD have treated the brain as a complicated, rather than complex, system. Complicated systems are composed of many parts, often intricately arranged; however, the assumption is that, overall, such systems tend to be linear, meaning their state and behavior follows clear, predictable patterns. Once the relationships between entities are understood—which could be done using statistical approaches for example—then the system's functioning can be explained or replicated.
We postulate that the brain (as a system) and its health (as its associated states and behaviors) are complex. Complexity refers to the intricate and dynamic interactions among components of a system, governed by numerous variables across multiple scales of space and time. Complex systems exhibit non-linear behavior, whereby small changes can lead to disproportionately large effects. They are high-dimensional, involving numerous interacting entities, and exhibit a probabilistic nature, when many processes are stochastic rather than deterministic, further complicating prediction. These systems also frequently feature adaptive mechanisms that make them difficult to predict using simple models, such as feedback loops, both positive and negative, which regulate processes and create interdependencies.
An inter-connected, plastic system at many scales
In our exploration of the brain as a complex system, we begin to disentangle the intricate nature of brain function and pathology by defining scales along a spatio-temporal continuum (Figure 1):
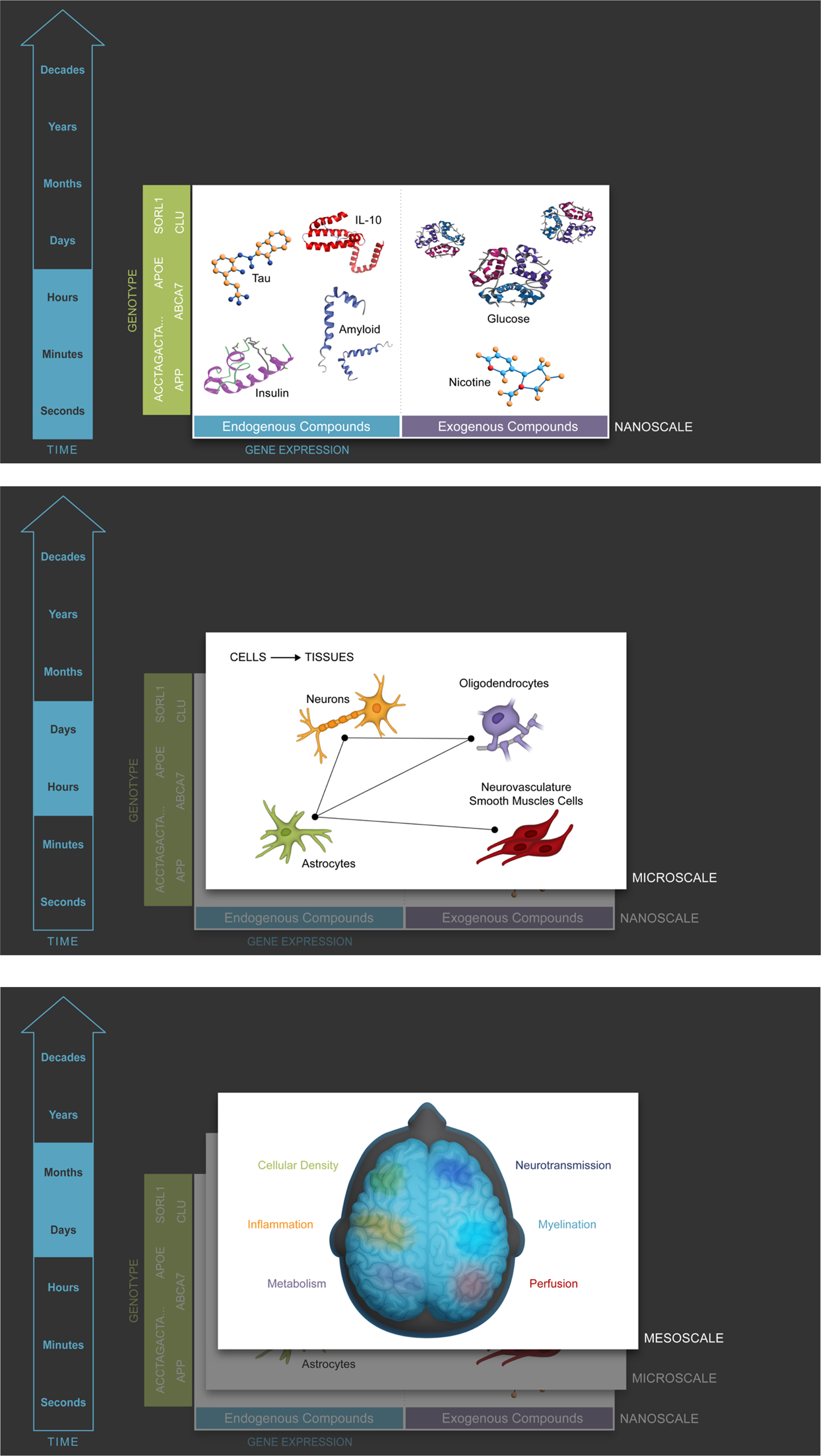
Multiscale framework of brain health and neurodegeneration. This diagram illustrates the interconnected complexity of brain health across multiple scales, from the nanoscale (genes and proteins), through the micro- (cells, tissues), meso- (structures), macro- (cognition, behavior), supra- (activities, interactions), peri- (dyadic relationships), to the exposcale (environmental context). Each scale represents a specific level of biological organization, contributing to overall brain function and pathology. Neurodegeneration is proposed to result from integrity failures across these scales, with disturbances at one level potentially cascading to others. The figure emphasizes the importance of a holistic approach to understanding and treating neurodegenerative diseases, such as Alzheimer's disease, through personalized interventions targeting the affected scales in each individual.
Nanoscale (genes and proteins)
Genetic expression and protein formation occur at the nanoscale, driven by genotype. For example, studies of Aβ plaque deposition or tau tangles give insights into the molecular underpinnings of neurodegeneration 4 whereas mutation in the APOE gene elevates the risk of AD. 44
Microscale (cells and cellular assemblies)
This scale involves the functioning of individual neurons and glial cells, as well as their organization into circuits. For instance, neuroinflammation, driven by the activity of microglia, and astrocyte dysfunction contribute to neurodegenerative diseases. 29 This level is also concerned with synaptic health and neurogenesis, which are crucial for maintaining cognitive function.
Mesoscale (regional brain structures)
The mesoscale examines larger brain structures, typically assessed through neuroimaging. For example, changes in hippocampal volume or glucose uptake in regions like the posterior cingulate cortex are indicative of neurodegeneration. 15
Macroscale (cognition)
At the macroscale, we consider high-level brain functions, such as memory, language, and executive functions. These functions emerge as the coordinated output of synchronized neuronal networks. Neurodegenerative diseases manifest here as cognitive impairments, which clinicians measure through neuropsychological testing. For example, individuals with AD often experience episodic memory loss before more global cognitive decline. 45
Suprascale (actions and interaction with the environment)
The suprascale relates to how an individual engages with their environment. This includes daily activities, social behaviors, and their ability to adapt to life challenges. For example, decreased social participation or difficulty managing activities of daily living is typically observed in dementia. 33
Exposcale (environmental context)
The exposcale focuses on the influence of environmental factors on brain health, both in terms of its physical (e.g., air pollution) and social environments (e.g., public policies). Long-term exposure to these factors has been linked to cognitive decline and neurodegeneration. For instance, exposure to high levels of air pollution has been associated with an increased risk of developing dementia. 33
These scales offer a comprehensive approach to understanding brain health and neurodegeneration, providing a complex framework to study how changes at one level may influence others. AD can be observed and analyzed at each of these levels, providing a comprehensive understanding of its multifaceted nature.
Across these scales, a variety of cell types, each exhibiting distinct gene expressions and arranged in diverse architectures throughout the brain, are interconnected in dynamic networks to perform multiple functions and execute intricate tasks in ever-changing environments. This arrangement is typical of complex systems. Thus, this complexity may appear chaotic but is in fact not; each element is subject to strict rules of interactions. It is simply that the multiplicity of interactions makes the output hard to predict without a clear understanding of these elements, rules, and relationships. This makes it ineffective to attempt to predict the future state of a complex system with reduction to a single factor. Such dimensional reduction is common in statistical analyses of AD (and elsewhere), where complex, multi-scale interactions are often flattened into simplified associations between isolated factors. However, the greater the reduction across scales—from nanoscale biology to exposcale environmental context—the weaker the ability to infer direct causality. A clear example is the observed association between lifelong exposure to environmental pollutants and the risk of AD: this relationship is likely mediated through multiple intervening mechanisms, and may also reflect broader determinants of health, such as socioeconomic status or access to care.
The dynamic nature of brain health states at each of the conceptual levels we have introduced is emblematic of complex systems. Consequently, any single-factor causal hypothesis or theory is by virtue of the complexity insufficient to explain the apparent heterogeneity and randomness. This diversity is, in fact, the manifestation of a complex system evolving into multiple instance states. While some commonalities can be identified, they are likely attributable to probabilities; the law of large numbers suggests that the aggregate effect of numerous non-normally distributed intermediate steps will approximate a normally distributed outcome.
This complexity underscores the necessity of considering the interplay of multiple factors to gain a holistic understanding of brain health and disease states.
Factors of brain health
To approach this complexity and make it amenable to mathematical modelling, let us identify factors that bridge brain entities across spatio-temporal scales and that appear necessary for optimal function.
Neuronal integrity
Neuronal integrity refers to the maintenance of healthy neurons, which includes the preservation of the neuronal structure, effective synaptic connectivity, and ongoing neurogenesis. Regarding the former, tau proteins are crucial for the cytoskeleton of neurons by stabilizing microtubules. In AD, tau tend to detach from microtubules and aggregates, eventually blocking transport and disrupting neuron signaling. Such neuronal damage can lead to neurodegeneration; and extensive neuronal loss has been amply recorded in AD, alongside synaptic dysfunction, due to several regulated cell death processes. 46
Myelin integrity
Myelin sheaths insulate axons, ensuring rapid signal transmission between neurons. The breakdown of myelin, known as demyelination, leads to slower neural communication, desynchronization across networks, and deficits in cognitive and motor functions. Myelin integrity is also crucial in neurodegenerative diseases like AD, where white matter degradation has been associated with cognitive decline. 47
Neurotransmitter integrity
Neurotransmitters like acetylcholine, dopamine, and serotonin are critical for communication between neurons. In AD, there is a marked reduction in acetylcholine, a neurotransmitter involved in memory and learning. Imbalances in neurotransmitters such as glutamate, GABA, and dopamine also contribute to cognitive deficits and neuropsychiatric symptoms in AD. 48
Metabolic integrity
Neuronal metabolism relies on glucose, lactate, and ketones to produce energy. In neurodegenerative diseases like AD, glucose metabolism is impaired, contributing to neuronal energy deficits and oxidative stress, while ketogenic processes appear maintained. Mitochondrial dysfunction also plays a critical role in the progression of neurodegeneration. 49
Cerebrovascular perfusion integrity
Adequate blood flow to the brain is essential for delivering oxygen and glucose, which neurons need for energy. Reduced perfusion, as seen in conditions like vascular dementia or AD, leads to ischemia and hypoxia, causing neuronal damage. Studies using imaging techniques like fMRI and PET have shown that cerebrovascular dysfunction occurs early in AD and is associated with cognitive decline. 28
Brain-blood barrier (BBB) integrity
The BBB protects the brain from harmful substances and maintains homeostasis. In neurodegenerative conditions, BBB dysfunction allows toxic substances, inflammatory agents, and immune cells to enter the brain, contributing to neuroinflammation and neuronal damage. BBB breakdown has been observed in early stages of AD and is thought to exacerbate amyloid-beta accumulation and tau pathology. 25
Immunological response integrity
The brain's immune response is mediated by cells like microglia and astrocytes, which are responsible for clearing debris, responding to injury, and protecting against pathogens. However, chronic neuroinflammation, often due to prolonged activation of microglia, is a hallmark of neurodegenerative diseases like AD. This sustained immune response can lead to neuronal damage and exacerbate disease progression. 29
Neurodegeneration as ongoing entropy
Failures at nano to suprascales
A multi-scale, multi-dimension framework can explain the wide-ranging nature and heterogeneity of dysfunctions and presentations which are partially captured in clinical definitions of dementia and AD. Integrity failures in any one or more dimensions can disrupt the entire system's homeostasis, leading to subsequent failures and resulting in a cascading effect over time. However, the multitude of potential failure modes at each scale complicates efforts to track, comprehend, and communicate these dynamics. Attempts such as the tri-dimensional ATN framework 6 can only represent eight (23) different states; with seven dimensions, one can account for 127 possible failure combinations (sum of the binomial coefficients), underscoring the complexity and interconnectedness of brain health, and very close to what is seen in practice in the study of Boyle et al. 50 The latter would suggest a mid-level dimensional state (±10E01), which would follow a power law in terms of statistical occurrence.
As we presented, a multiplicity of genes and proteins have been correlated with disease expression at the nanoscale, 51 while at the cellular level (the “microscale”), a host of issues that plague nearly all brain cell types have been found, with processes extending from hours to days or months. 52 At the organ level (the “mesocale”), these alterations aggregate over years in multiple brain regions to disturb both anatomy 53 and physiology, 54 eventually affecting cognition and behavior (the “macroscale”) on decades-long timeframes (Figure 1) in patterns both common and irregular. Hence, from an initial state that is primarily determined by genetics and resilience driven by lifetime events, decline trajectories are formed through the interplay of a variety of insults and repair mechanisms, interactions, and iterations between dimensions at these different spatio-temporal scales.
Rather than additive, failures should be seen as multiplicative, as a failure of any one component in any one conceptual layer will have reverberating impacts. When complex systems evolve iteratively through time, effects cascade through to engulf the entirety of the system. It then becomes nearly impossible to ascertain what was the single etiological source; rather, several systems become affected. By then, uncovering the initial source of the cascade becomes rather academic; multiple dysfunctions must be corrected to retain, or even regain, some form of homeostasis. Conversely, stressing unitary resilience is not sufficient to guard the whole system from eventual decay.
Commonalities and differences in entity failure impacts
It is important to note that failures will be unequally distributed in both time and space, with important consequences. Entropy, acting on biological systems as it does on any other, increases over time and will lead to the progressive accumulation of failures. For biological systems, this could be considered the baseline senescence related to aging. In addition to that, we engage in behaviors and accumulate through lifestyle additional stressors that apply further pressure on systems, increasing the rate of failure. At some point, the system's output has diminished sufficiently to be qualified as having failed—the loss of integrity.
In engineering terms, this concept is referred to as mean time between failures (MTBF). In human-made, artificial constructions (an airplane, a bridge), sub-systems are designed to be both redundant and repairable to ensure that the whole system can continue functioning past an individual component's MTBF. Similarly, nature has endowed brain health factors with growth/repair/flexibility mechanisms exhibiting the same behavior. Yet, upon reaching their MTBF, either the primary brain health factor or its associated repair mechanism will enter an integrity failure zone in terms of functional outputs; one could qualify this as a lack of plasticity (albeit of a multi-factorial nature, not simply cognitive 30 ). Hence, if we assume the distribution of MTBF among brain health factors to be normally distributed, this implies that some of them will fail more frequently than others. Meanwhile, these entities are not expressed uniformly across the brain, based on gene expression, cellular organization, and functional requirements. Given the networked nature of brain function, the interaction between these two dynamics therefore implies a non-uniform impact of failure for each dimension that would be quite specific to an individual brain instance. However, when viewed from the population level, the central limit theorem would dictate that we end up with a distribution of negative outcomes where certain regions/dimensions of the brain fail more often than others for specific reasons. This non-uniform distribution will therefore present patterns, which could be misconstrued as specific diseases; rather, they are but the most likely modes of probabilistic failure.
For example, one of these common modes of failure could implicate neuronal and perfusion integrity failures in the hippocampus, a region poorly served in vessels while subjected to high cognitive load 55 ; the resulting functional impact, related to memory, would be seen as being a central element of the most common neurodegenerative disorder (in this case, AD). Yet neuronal and perfusion integrity failures do happen in other brain areas, with similar tissue impact; however, with a lower probability—and with a functional outcome that is clinically different, and often reported as such (e.g., visual variant of AD 56 ).
Having a multifactorial, multiscale theoretical framework provides a novel perspective on the etiology of neurodegeneration on the AD spectrum, emphasizing the need to consider the brain as an intricate and dynamic system subject to the principles of complex systems.
Reconciling disease definitions from different scales
Researchers seem bent to define AD depending on which scale they work in: public health practitioners at the expo- and suprascales; clinicians at the macroscale; pathologists at the microscale; and bioscientists at the nanoscale. Besides sociological bias, since effects are varied, this creates an ill-posed problem: continuity is not assured between scales.
It also follows that defining diseases based on failures at any one scale is not an approach that can lead to finding the underlying causes of dysfunction, as there exists a host of possibilities that can lead to that outcome. For example, focusing on a single mechanism at the nanoscale layer will fail to explain the multiplicity of cognitive and behavioral presentations that will arise after several years, once multiple systems are affected as the impact is felt through the complex system, missing the forest for the trees. Meanwhile, adopting a symptoms-based definition at the macroscale has outstanding value in clinical practice; however, in its purest form it cannot adequately address the underlying causes of these symptoms and misses the trees for the forest. Rather, a multi-layer and multi-dimensional formulation opens the field of neurodegeneration to correctly understand the major, cross-scale effects that are present for any one patient, thereby allowing for the tailoring of therapies to address the situation in its globality.
Identifying neurodegeneration entities in their most functional, biological elements at the nano- to meso-scales does not negate their differential impact clinically. What we claim here is that attempting to define the problem by mixing scales only creates confusion. A loss of integrity anywhere in the brain is a loss of integrity, should be recognized as such, and treated accordingly. The case of amyloid deposition is perhaps the most blatant example of this. No sooner had the field determined that AD corresponded to Aβ deposition (a nano/microscale concept) that the definition was commandeered to attempt the grafting of information from other scales to explain the myriad presentations seen in the clinic: “typical” AD when deposition occurs in temporo-frontal areas and “atypical” elsewhere; “tauopathic” when the latter protein was present or not, and so forth. This is the field's attempt at reducing a high-dimensional problem into fewer, more manageable entities; there is a limit to this process.
A redefinition of neurodegeneration based on brain health
We propose to embrace this complexity rather than attempting to simplify it. Current diagnostic criteria for AD have predominantly been descriptive, focusing on identifying the most common and statistically significant markers, such as Aβ plaques, tau tangles, and specific cognitive impairments. These criteria are designed to categorize and diagnose based on observable symptoms and biomarker presence but fall short of providing an explanatory framework for the underlying mechanisms.
This descriptive nature means that diagnostic criteria often lag new discoveries, such as the identification of TDP-43 pathology or the integration of biomarkers from clinical and pathological studies. As a result, the field must continuously update and reconcile diagnostic criteria with emerging research to accommodate new entities and varied viewpoints, as the combinatorial nature of pathologies 50 increases with each new discovery. This gap underscores the necessity for a theoretical framework that not only describes AD but also explains the multi-dimensional and multi-scale processes driving its progression, the co-occurrence of multiple dysfunction at the same time, and the ability to incorporate additional findings without necessitating a complete overhaul.
Our proposed framework supports the perspective of neurodegeneration as a syndrome, with entity integrity as etiologies. The major challenge in the field, defining exactly the presentations at each scale of entities such as AD, dissipates when we recognize it as a multiscale problem with varied presentations and manifestations at each level. This variability, which differs from one individual to another, allows for a more nuanced understanding and approach to treatment.
Resilience, redefined
A multifactorial, multiscale viewpoint further leads to a subtle reinterpretation of “resilience” 57 as an emergent property of parameter dynamics across time and scales, rather than a static or latent capacity. Specifically, brain health in adulthood is set according to the expression of neurobiological parameters leading to neuronal, connectivity, or cerebrovascular integrity. These parameters however evolve when adapting or recovering to internal or external perturbations across the life course, mediated by molecular, cellular, behavioral, and environmental influences, whether they are voluntary (e.g., positive or negative health behaviors) or involuntary (e.g., exposure, social determinants). Diverging with the classical interpretation, brain maintenance, brain reserve, and cognitive reserve, rather than standalone latent traits or proxy measures, are better understood as the observed consequences of these parameter trajectories, at respective scales: the measurable functional output resulting from the dynamic interplay of entities within the complex, multi-scale brain system. In this formulation, resilience is not an intrinsic variable, but a state-dependent outcome shaped by the probabilistic evolution of integrity across brain systems over time, in interaction with life-course exposures and context. This view aligns mechanistic modeling with epidemiological observations and dissolves artificial boundaries between constructs that have historically been siloed. It also suggests more direct means of interventions to support functional outcomes in the face of neurodegeneration.
Implications for therapy
Therapy choices as viewed from different scales
Therapeutic choices depend significantly on the scale at which the intervention is targeted, e.g., macroscale interventions include cognitive stimulation and training, while nanoscale interventions target protein misfolding and sub-cellular processes.
When viewed as a complex system, a comprehensive, whole-brain effort is the only option that is likely to bear consistent and durable fruit. As neurodegeneration progresses and multiple factors approach failure, increasing energy must be expanded to return the system to some form of homeostasis. At earlier times, it becomes crucial to identify the primary modes of failure that are present in any one individual to tailor effective therapeutic strategies. By understanding the specific scales at which disturbances occur, clinicians can design more personalized and effective interventions, improving outcomes for patients. This multiscale approach not only acknowledges the complexity of the neurodegenerative process but also leverages the strengths of various specialties to address the multifaceted nature of brain health.
Evidence to this effect can be glimpsed by comparing mono- to multi-scale therapeutic approaches. Amyloid antibodies have had unquestioned success at achieving their desired targets (i.e., removing Aβ plaques) but limited efficacy otherwise at the macro- and suprascales, putatively due to a dampening of effects as it cascades up to the level of cognitive outcomes, alongside other (deleterious) pathways that remain unaffected by the therapy. Physical exercise, on the other hand, is known to affect multiple systems at multiple scales by affecting gene expression, molecular releases, cellular processes, cerebrovasculature, and cognition.. 58 Perhaps unsurprisingly thus, exercise was shown to achieve, in a wide array of patients, effect sizes that surpassed pharmacological approaches. 59
Hence, therapeutic interventions that target an entity at a specific scale can be viewed to permeate and affect only that scale, with the timeline for changes corresponding to the period of expected alterations at that level. For instance, amyloid therapies demonstrate this principle clearly: they achieve amyloid clearance at the mesoscale within weeks, but cognitive improvements at the macroscale take months to manifest, while being of a much lower magnitude that amyloid removal. 17 This disparity underscores the importance of understanding the distinct timelines and interactions across different scales.
It is crucial to recognize that dysfunction is not solely a bottom-up process, where issues at the nanoscale propagate to the scales “above”. Rather, there is significant downregulation, with interactions occurring abundantly between scales. This bidirectional influence suggests that combinatorial therapies, which address multiple scales simultaneously, are likely to be the most effective.
In general, there will be a strong confirmation bias in play when mono-therapies targeting a dimension at any given spatio-temporal scale are judged based on their achieving an outcome within the same scale. This will lead to an over-estimation of the efficacy of the intervention, as the disease should be viewed as a function of cross-scale brain health status. Global outcomes should thus be prioritized and incorporated in the design of future therapeutical trials.
Preventive approaches
As entropy increases within the system, it becomes increasingly challenging to restore homeostasis. Preventative measures are thus best viewed as maintaining this homeostasis, while therapeutic interventions aim to re-establish balance in a system already showing signs of strain.
Epidemiological studies show that risk factors for dementia vary with age and cumulative exposure over time. The twentieth century saw significant shifts in lifestyle, healthcare, and environmental conditions, which have influenced dementia risk. Reduced exposure to certain risk factors correlates with a decline in the incidence of dementia.60–62
Multi-factorial interventions, addressing various risk factors simultaneously, have the best chance of mitigating dementia. Supporting this hypothesis are interventions like exercise programs and the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER), 63 which target multiple dimensions across different scales. These interventions have shown effectiveness in reducing cognitive decline by promoting cardiovascular health, cognitive health, and overall well-being.
Incorporating suprascale factors into our multi-scale, multi-dimensional framework underscores the importance of addressing dementia through a broad, preventative approach. Recognizing the cumulative impact of risk factors over a lifetime and implementing comprehensive interventions can effectively reduce the burden of dementia. This perspective aligns with the growing body of evidence supporting the effectiveness of lifestyle modifications and multi-factorial strategies in promoting brain health and delaying dementia onset. Extending this logic, the promotion of brain health, i.e., the earliest adoption of life-long strategies that are beneficial, should be an even better approach, leading to the reduction of the incidence of the risk factors themselves.
Clinical trials
Randomized controlled trials (RCTs) are based on the premise that, all other things being equal, the exposure will result in a statistically significant difference in a single, primary outcome. While there may be such as a thing as an “average liver function” for a group of individuals representative of a population, RCTs as we know them for brain disorders as complex as dementia are bound to fail.
The evolution of outcome definitions primarily drives this shift. Initially, the focus was at the suprascale to periscale level, such as placement or institutionalization. Gradually, it has transitioned down to instrumental activities of daily living and then to cognition at the macroscale. Currently, it encompasses a mix of micro and mesoscale factors, such as evidence of plaque removal across the brain as observed via PET scans. The trend is now moving towards the nanoscale, focusing on the removal of Aβ as detected in cerebrospinal fluid or blood, an imperfect surrogate for clinical performance.
This shift introduces several challenges. Firstly, not everyone accepts the new outcomes, as disease definition tends to follow professional areas of expertise. Secondly, a nanoscale outcome for a single factor may not necessarily translate to a meaningful change at the macroscale; so far the evidence is clear on this point. It is highly probable, but remains to be convincingly demonstrated, that multiple dimensions must be influenced simultaneously to achieve a significant impact across all scales. Conversely, the specificity of a macroscale or suprascale change is often too broad to be attributed to any single nanoscale or microscale change. The complex nature of the brain makes pinpointing a single cause extremely difficult.
Secondly, most clinical outcomes also chronically underestimate the intra- and inter-rater variability associated with the instruments. For example, the intra-rater variability of the CDR SOB (87% according to Wolf et al., 64 which, transposed on the scale, represents 0.9 points) is superior to the magnitude of the change observed in the trial (confidence interval of 0.23 to 0.67, according to Van Dyck et al. 17 ); a similar observation can be done for all scales in all -umab trials. This alone should give us pause.
Thirdly, the timescales for observing changes differ significantly. For example, a change rate of 1% per day can yield a statistically significant change of 20% at the nano-micro scale within less than a month. However, if the change rate extends to weeks, achieving the same result would take approximately half a year (experienced by acetylcholine inhibitors 65 ). When the rate is measured in months, it aligns with a 1.5 to 2-year follow-up period as observed in the latest -umab trials. This discrepancy in timescales is an inevitable consequence of the scale at which outcomes are measured.
Finally, traditional statistical methods may fall short. The current standard in RCTs is to test group differences in a single dimension with a certain power, effectively compressing all entities, each with their own parameters, into one dimension. This is exceedingly difficult to achieve. For statistical analyses to be meaningful, they must be confined within a single scale or between “adjacent” scales. For instance, studying amyloid clearance at the nanoscale should be directly related to deposition at the micro/mesoscale, rather than to changes in brain structure or function at the macroscale. Ensuring temporal and spatial congruence between scales is essential for making valid causal inferences, especially in the absence of a clear and robust functional model of brain health.
Multi-scale, multi-dimensional hypotheses
As we posit a multi-scale, multi-dimensional framework for assessing brain health, its likelihood increases if the following hypotheses are proven correct:
Take a step back
Dysfunctions at the entity level are distributed normally. Given the specialization of brain regions however, their effects will differ. Hence, at the level of populations, we should see clinical phenotypes emerge as a function of large population statistics in proportions like epidemiological data.
More is better
Targeting multiple dimensions across different scales should be more effective than targeting any one dimension at any one scale.
Many fits all
Individuals age and their brain health deteriorates in patterns that are specific to them. Thus, interventions must be personalized in the generic sense, i.e., specific targets for specific scale states should be considered. The same targets should not be as effective for two individuals with different dimensional states.
Magic is not allowed
When altering a dimension at a given scale, the effect should be trackable to its next adjacent scale. Effects cannot jump scales.
There is no free lunch
Conversely, if an effect occurs at a different scale than the one targeted, it implies a hidden or indirect relationship between dimensions.
Special powers do not exist
There is no need to rely on concepts that cannot be incorporated (“made into flesh”). They are but the expression of states of entities at all scales, performing their functions, for any given moment for that individual.
Computational models as a way forward
Computational models as an effective framework to test theories
To study these complexities, a new class of methods is required. Epidemiology lacks specificity regarding how factors, including risk factors, exert their effects on neurobiological pathways, especially at the level of a single individual. Mechanistic studies are overall designed to test one or few pathways in limited and homogeneous samples, and often on pre-clinical models that have limited relevance to humans. This makes them ill-suited to explain population level incidence and prevalence and to experimentally assess combinations of factors, as it quickly becomes logistically and statistically impossible due to the substantial number of variables involved. With regards to RCTs, studying patients with a multi-factorial approach faces ethical, logistical, and financial challenges, including the lack of causal studies, the long duration of neurodegeneration disease processes, and incomplete biomarker selection in clinical studies due to resource and time constraints. Finally, while data mining and machine learning can reveal valuable patterns and correlations in large datasets, 66 they face several limitations when attempting to uncover causal relationships in cohort data, particularly when these data have been acquired according to specific experimental paradigms and/or are subject to selection biases and confounds. Deep learning methods, in particular, excel in large, well-labeled datasets but often lack interpretability, particularly in complex biological systems.
Mathematical modeling on the other hand is a great tool to understand complex mechanisms such as dementia. Mathematical models provide a way to explore and conceptualize principles by merging biological knowledge with experimental data into model simulations. 67 In mathematical models, physical reality is abstracted into entities and parameters that can help us more easily understand relationships. Mathematical models are defined a priori; while their parameters may be learned, it is primarily their predictions that are subjected to validation against real data. Hence, if a model makes predictions that are out of line with observed results, or that cannot be verified experimentally altogether, either the entity relationships or assumptions must be modified to conform to reality.
In the case of neurodegeneration, mathematical models could help us figure out causal mechanisms and therefore propose targets for disease prevention. Accurate models should help with intervention planning and trial, by testing in silico the potential effect of different drugs. While nascent, the field is making interesting strides forward. In a recent review we conducted of 18 such models focused on AD, 68 we noted a strong preponderance of models at the nano/microscale, primarily focused on Aβ, and secondarily on tau; only one took a more integrative perspective that reached out to other known aspects of AD, such as inflammation and reactive oxygen species. 69 None so far address other scales, such as connectivity at the mesoscale, 70 necessary for cognition.71,72
Translating to real-world testing
We call for a concerted effort to utilize computational models as the primary method for testing these hypotheses quickly and efficiently in silico. Computational models can simulate the intricate interactions within the brain, providing valuable insights into how different factors contribute to the progression of neurodegeneration. These models can help identify potential therapeutic targets and predict the outcomes of various interventions.
If a computational model fails to produce the expected results, it prompts a re-evaluation of the underlying theory, allowing researchers to refine their hypotheses and improve their understanding of the disease. Conversely, if a model successfully predicts outcomes that align with real-world data, it provides a strong rationale for advancing to real-life testing.
Successful in silico findings should be translated into real-life applications through rigorously designed RCTs that track factors individually. Outcomes must be redefined within a multidimensional framework, employing relevant metrics. The focus should be on meaningful movement along various dimensions, which can be extrapolated using the covariance between these dimensions. This approach will provide a more comprehensive and accurate assessment of the outcomes.
Conclusion
Alzheimer's disease is a complex, multi-dimensional problem that manifests through various interconnected layers, each contributing to the overall pathology. This multi-scale nature encompasses genetic, molecular, cellular, structural, cognitive, and environmental factors.
The prior emphasis on unifactorial viewpoints is possibly an artefact of research, including the well-known bias towards positive research reporting, 73 which attracts both attention and funding, creating a self-perpetuating loop. Further, the necessity to provide results that have reached statistical significance will naturally be easier to achieve in limited experimental settings, given statistical power considerations, and thus favor the study of individual effects over the logistically much more difficult task of deciphering interactions in a system; complexity increasing factorially with each new entity and parameters being considered.
However, this unifactorial approach does not necessarily lead to an increase in understanding. Rather than shying away from complexity, we propose to embrace it. By examining integrity across multiple dimensions and scales, we can define, measure, and influence neurodegenerative processes with better efficiency. Such a comprehensive approach would allow for a deeper understanding of neurodegeneration's multifactorial nature. By leveraging computational models to capture this complexity, we could test hypotheses and efficiently identify and refine potential treatments. This iterative process, from in silico modeling to real-world testing, would drive the discovery of effective interventions, ultimately leading to better prevention, management, and treatment of neurodegeneration such as what we call today Alzheimer's disease.
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is supported by various operating grants from the Canadian Institutes for Health Research to S.D. (grant numbers PJT-469654, PJT-159778).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: SD: Officer and shareholder of True Positive MD Inc. Paid consulting for Eisai and Novo Nordisk. Unpaid consulting for Eli Lilly. OP: No conflict. CH: Paid rater in clinical trials of Eli Lilly and Bristol Myers Squibb. CB: Shareholder of Imeka Inc.