
Abstract
Background
Alzheimer's disease (AD) disrupts histone acetylation/deacetylation homeostasis, blocking access of transcription factors to DNA, and compromising learning. Vorinostat (VOR), the only FDA-approved HDAC inhibitor that is orally bioavailable and brain penetrant, confers neuroprotection in AD models. We delivered VOR via diet in an AD mouse model, examining tolerability, accompanied by biochemical analyses.
Objective
Our objective was to examine dietary delivery of vorinostat for tolerability, including changes to histone acetylation, amyloid-β (Aβ) production, oxidative stress (OS), mitochondrial health, and synaptic integrity.
Methods
Food pellets containing control, 0.18 mg/g (low-dose) and 0.36 mg/g (high-dose) VOR were administered to hAβ-KI AD mice for 14 days. Brain acetyl-histone H3 (AH3), total H3 expression, and synaptic markers were measured via Western blot. Aβ, H2O2, antioxidant capacity, lipid peroxidation (via 4-hydroxynonenal (4-HNE)), adenosine triphosphate (ATP), and citrate synthase (CS) activity were measured in brain tissue.
Results
VOR inhibited brain HDAC enzyme activity and increased AH3 and H3 expression at both VOR doses. Aβ and synaptic proteins were not significantly affected; however, OS markers were improved at both doses. Both doses increased CS activity, while ATP was increased only at the low dose. Finally, low-dose VOR was tolerable over 2 months.
Conclusions
We established that low-dose VOR, delivered via diet, is tolerable in AD mice, successfully inhibiting brain HDAC activity while reducing OS and improving mitochondrial health. This study improves existing preclinical experimental designs by enabling noninvasive manipulation of histone acetylation through dietary intervention. This route of administration provides advantages for future preclinical animal studies.
Keywords
Introduction
Histone acetylation, balanced by competing activities of histone acetyltransferases and histone deacetylases (HDACs), is an essential mechanism of epigenetic regulation. 1 There is growing evidence that Alzheimer's disease (AD) involves a disruption in histone acetylation/deacetylation balance. 2 AD patients exhibit aberrant acetylation of histone lysine residues.3,4 Several studies support upregulation of a subset of HDACs in AD.5–7 Adult hippocampal neurogenesis, which is highly dependent on epigenetic regulation of histones, is deficient in AD.8,9 Recently, using a secondary analysis of a postmortem AD transcriptomic dataset, 10 we found that HDACs 1, 4, and 7 were upregulated in the hippocampus of AD patients relative to age-matched non-AD individuals. 11 Moreover, non-histone proteins, such as calmodulin, are deacetylated in human AD. 12
Preclinical evidence also supports HDAC dysregulation. Hippocampal HDAC3 was increased in APP/PS1 AD mice; its depletion improved spatial learning and reduced amyloid-β (Aβ) levels. 13 Moreover, the genetic deletion of HDAC3 improves long-term memory formation in non-AD mice. 14 Diminished H3K9 acetylation of AD-related genes, 15 reduced histone H4 acetylation, 16 and reduced histone H9 acetylation 12 were shown in AD mouse models. Mechanistic evidence supports close bidirectional relationships between HDACs and amyloidosis.17,18 Beyond the classical HDACs (classes I, II and IV), there is also evidence that the dysregulation of sirtuins (class III HDACs), and SIRT1 in particular, in AD exacerbates neuroinflammation and Aβ aggregation. 19 While sirtuin expression is diminished rather than increased in AD, 19 this further highlights epigenetic changes as causative factors in AD progression. Taken together, this human and preclinical evidence converges on HDAC dysregulation in AD, implicating HDACs as logical targets for AD prevention and/or treatment strategies.
Vorinostat (VOR, or SAHA (suberoylanilide hydroxamic acid), brand name Zolinza), is a class I/II HDAC inhibitor (HDACi). Of four US FDA-approved HDAC inhibitors, only VOR is orally bioavailable and crosses the blood-brain barrier (BBB).20,21 Although originally FDA-approved for cancer, HDACis have received attention for AD and other neurodegenerative disorders.21–24 Clinical observations point toward a potential neuroprotective use of VOR. 25 In preclinical AD models, VOR normalized deficits in hippocampal γ oscillations and interneuron firing when applied to hippocampal slices from PS/APP AD mice. 26 Moreover, VOR in drinking water restored cognitive function in APP/PS1-21 AD mice, which was accompanied by anti-inflammatory effects and restoration of transcriptional control. 27 Other HDACis have also demonstrated promise in AD models.28–34 However, sirtuins are unaffected by classical HDACis, 35 including VOR.
To date, most preclinical studies using VOR in mouse models with behavioral analysis have administered VOR at doses ranging from 5.0–50 mg/kg, typically by intraperitoneal (IP) injection, for limited durations ranging from 2–7 weeks.36–41 However, it is unclear whether such short-term interventions can substantially ameliorate the neurodegenerative progression in AD models. Additionally, VOR administration via IP or oral gavage (OG) may suffer from practical limitations. First, repeated IP injections cause injection site pain/scarring. Second, short-term IP/OG dosing lacks demonstrated tolerability for long-term treatment strategies. Third, accidental user-induced injury via OG or IP may increase the attrition rate, which is already substantial in some AD mouse models. Finally, with OG and IP, the labor cost is significant, particularly for long-term treatments.
In this study, we tested a dietary formulation of VOR using the humanized Aβ knock-in (hAβ-KI) late-onset AD mouse model. 42 Characteristics of the model include age-related increases in insoluble Aβ, formation of hippocampal Aβ protofibrils with an absence of plaque formation, reduced expression of synaptic proteins, and the emergence of behavioral deficits by 10 months of age. 42 More recent work has reported reduced expression of mitochondrial biogenesis genes, disrupted mitochondrial dynamics, and behavioral deficits at 7 months of age. 43 We determined that a low dose of VOR, delivered via diet, inhibits brain HDAC enzymatic activity, increases brain histone acetylation, and is tolerable over an extended time frame (months). Since oxidative stress (OS) and mitochondrial dysfunction are thought to play a role in AD progression, 44 we examined H2O2 levels, total antioxidant capacity (TAC), lipid peroxidation via the lipid peroxidation product 4-hydroxynonenal (4-HNE), citrate synthase, and adenosine triphosphate (ATP) levels. We found that a low dose of VOR improved OS measures indicative of neuronal health, and these effects were more pronounced in males than females. We conclude that VOR can be effectively delivered via diet to achieve both safe and potentially suitable dosing for long-term AD prevention studies.
Methods
Mice
Homozygous hAβ-KI mice (Strain# 030898) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). These mice possess a ‘humanized’ APP gene, yielding an Aβ peptide sequence that is identical to humans. 42 Unlike APP/Aβ overexpressing AD models, 45 hAβ-KI mice express APP at normal physiological levels, better resembling human AD progression. 42 Mice were housed in groups of 2–3 and given ad libitum access to water and food in standard shoebox-style caging. Food consisted of either 5R53 chow (Cat# 3002890-748, LabDiet, St Louis, MO, USA), AIN-93M purified diet (DYET# 105147, Dyets, Inc., Bethlehem, PA), or AIN-93M supplemented with VOR at 0.18 mg/g or 0.36 mg/g (DYET# 105219 and DYET# 105150, respectively, Dyets, Inc.). Mice were housed at an ambient temperature (22 ± 2°C) with 12-h light/- dark cycles and humidity of 45 ± 10%. Brains were extracted and hemisected. The left hemisphere was snap-frozen in liquid nitrogen and stored at −80°C until used.
Ethics statement and regulatory compliance
This study was carried out in accordance with the recommendations of U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the TTUHSC Institutional Animal Care and Use Committee (TTUHSC IACUC protocol numbers: 22012 and 22013). Efforts were made to minimize pain and suffering. Experiments for the study were performed using laboratories, equipment, and safety procedures compliant with the TTUHSC Institutional Biosafety Committee (IBC# 16008).
VOR diet formulation
VOR (SAHA, MW: 264.32) was purchased from Tocris (Cat# 149647-78-g; Minneapolis, MN, USA). AIN-93M purified diet was produced with VOR contents of 0.0 (control), 0.18, and 0.36 mg/g of diet. AIN-93M represents the most up-to-date dietary recommendations from the American Institute of Nutrition. It was formulated in 1993 as an updated replacement for the AIN-76 and AIN-76A diets.46,47 Gamma-irradiation was not used to reduce bacterial load. Purified diets naturally have lower bacterial loads than chows, 48 and it has been reported that gamma irradiation destroys a considerable portion of VOR in nanoparticles. 49 Irradiation also destroys certain nutrients, such as vitamins A and B6, as well as greatly increases peroxide levels. 50 VOR was reported to be stable in food for >30 days. 51 The diet with 0.36 mg/g VOR content was intended to target a weekly drug delivery of 250 mg/kg mouse body mass, which is equal to the total weekly drug administered if dosed for 5 days/week at 50 mg/kg by OG or IP injection. Due to potential tolerability concerns, we also tested an identical AIN-93M-based diet with a reduced VOR content of 0.18 mg/g. All drug content calculations accounted for the fact that with dietary administration, the target dose would be consumed over 7 days per week rather than administered via OG/IP over 5 days per week.
Dietary dosing of VOR in hAβ-KI mice
Doses of VOR were chosen in which behavior was tested in mice.36–38 To assess VOR dose in diet over two weeks, mice (10–12 months of age) were fed an AIN-93M purified diet with 0 mg/g, 0.18 g mg/g or 0.36 mg/g VOR, each involving 5 male and 5 female hAβ-KI mice with ad libitum access to food and water for 14 days. Mouse and diet weights were measured at days 0, 7, and 14. Mice were monitored daily for any adverse drug effects. After 14 days of treatment, mice were sacrificed, brain tissues were collected and analyzed for acetylated histone H3 and histone H3, HDAC enzyme activity, Aβ, H2O2, total antioxidant capacity, 4-HNE adducts, citrate synthase activity, ATP, and synaptic proteins. Because there were multiple mice per cage, the consumption for each mouse is a weighted estimate based on a mouse's body weight relative to the total body weights of all mice in the cage. To assess VOR over two months, 14–17-month-old hAβ-KI mice (6 females and one male per group) were used. Body weights were tracked for individual mice weekly for 9 weeks, and diet was added weekly. One female on the control diet had to be euthanized 4 days into the experiment due to ulcerative dermatitis, and one female on VOR diet was found dead 22 days into the experiment. These animals were excluded from the analysis.
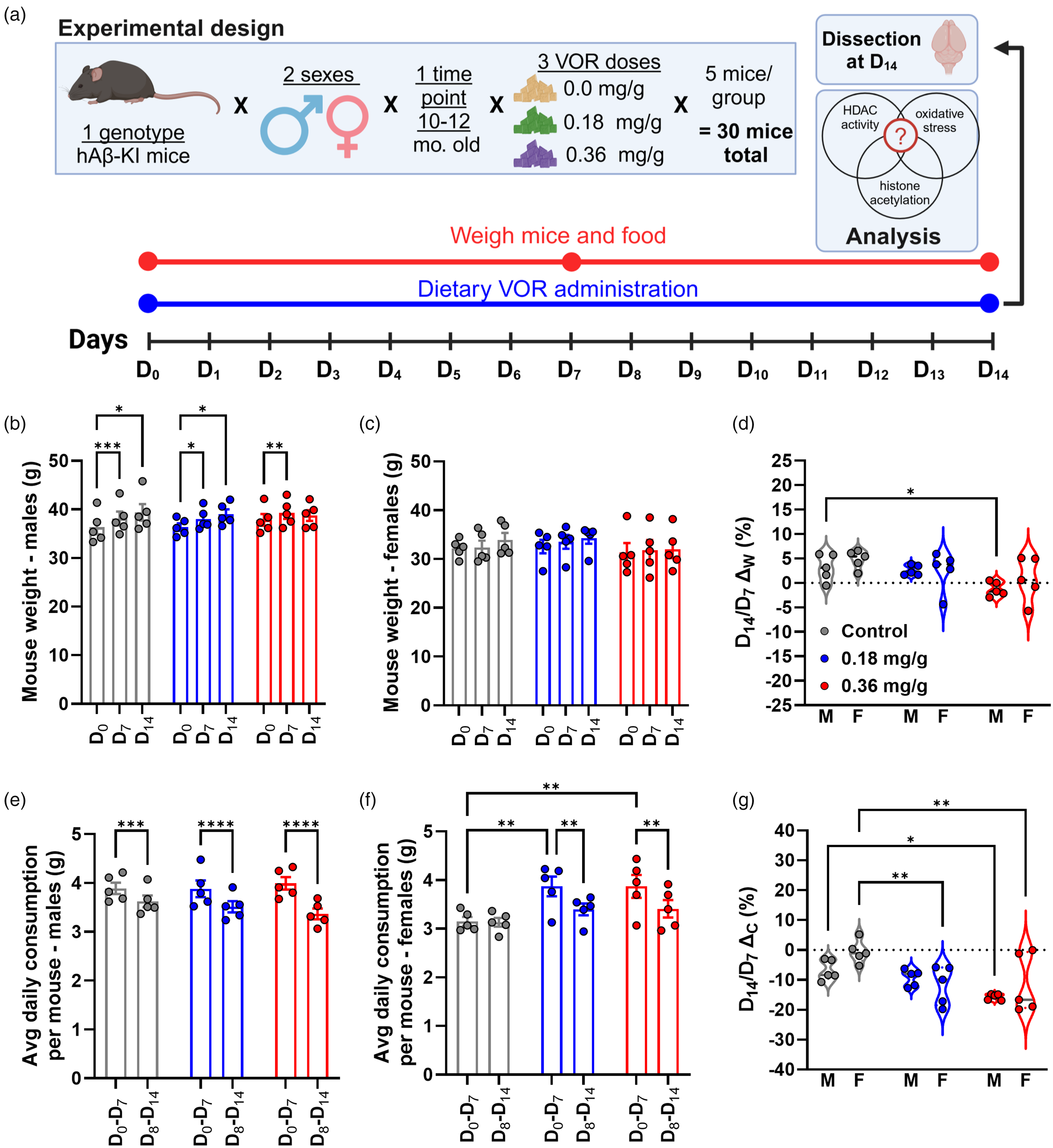
A low dose of dietary VOR was tolerable for 14 days. (a) Experimental design to test the tolerability of AIN-93M diets formulated with VOR. Body weight for (b) males and (c) females across days 0 (D0), 7 (D7) and 14 (D14). For b–g, treatment groups are denoted by colors: control (0 mg/g VOR, gray), 0.18 mg/g VOR (blue), and 0.36 mg/g VOR (red). (d) Percent weight change, as a ratio of D14/D7, across treatment groups and sex (M: male; F: female). Average daily diet consumption during D0-D7 and D8-D14 of treatment, for (e) males and (f) females. (g) Percent change in diet consumption, as calculated as a ratio of D14/D7 across treatment groups and sex (M: male; F: female). Statistical significance is denoted by asterisks: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Western blot (WB) analysis
AH3 was assessed as a marker of HDAC inhibition, as described previously. 52 Tissue and cell lysates were prepared in 1X RIPA buffer containing protease inhibitors. Protein extract was quantified using Pierce 660 nm Protein Assay Reagent (Cat# 22660, ThermoFisher Scientific, Waltham, MA, USA). Total cell lysates (45 µg/lane) were loaded on 4–12% bis-tris gels (Cat# WG1402BOX, ThermoFisher Scientific) in MES gel running buffer (Cat# NP0002, ThermoFisher Scientific). Proteins were transferred to nitrocellulose membrane (Cat# 1620097, Bio-Rad Laboratories, Hercules CA, USA) using Bolt Transfer Buffer (Cat# BT00061, ThermoFisher Scientific). Nonspecific binding was blocked using 5% Blotto nonfat dry milk (Cat# sc-2325, Santa Cruz Biotechnology, Dallas, TX, USA) with 0.1%Tween-20 for 1.5 h at room temperature (RT). Membranes were probed with rabbit monoclonal anti-acetyl-Histone H3 (Lys9, C5B11, Cat# 9649, Cell Signaling Technology, Danvers, MA, USA) diluted 1:1000 in 5% BSA in Tris-buffered saline with 0.1%Tween-20 (TBST; 20 mM Tris·HCl (pH 7.6), 137 mM NaCl, and 0.1% (vol/vol) Tween-20), rabbit monoclonal anti-Histone H3 (D1H2, Cat# 4499, Cell Signaling Technology) diluted 1:2000, mouse monoclonal anti-synaptophysin (SY38, Cat# ab8049, Abcam, Cambridge, UK) diluted 1:700, mouse monoclonal anti-PSD-95 (6G6-1C9, Cat# NB300-556, Novus Biologicals, Centennial, CO, USA) diluted 1:1000, in 5% nonfat dry milk in TBST, and incubated overnight at 4°C using gentle shaking. Membranes were washed with TBST 3x 5 min each and incubated with horseradish peroxidase-conjugated anti-rabbit IgG (1:2000 dilution, Cat# 7074, Cell Signaling Technology) and anti-mouse IgG (1:2000, Novus Biologicals), for 1 h at RT in 5% non-fat dry milk in TBST. For visualization of the bands, Super-Signal West Pico PLUS Chemiluminescent Substrate (Cat# 34580, ThermoFisher Scientific) was used, following the manufacturers’ instructions. For the loading control, at the end of the experiments, nitrocellulose membranes were stripped with WB stripping buffer (Cat# sc-281698, Santa Cruz Biotechnology) and re-probed with rabbit polyclonal antibody for β-Actin (1:1000 dilution, Cat# 4967, Cell Signaling Technology). Bands were visualized using an ImageQuant LAS4000 (GE Healthcare Life Sciences, Chicago, IL, USA) and were quantified using ImageJ FIJI (v1.54f) software. 53
Nuclear extract preparation
To prepare nuclear extracts, EpiQuik Nuclear Extraction Kit II (Cat# OP-0022-100, Epigentek, Farmindale, NY, USA) was used as directed by the manufacturer, with minor modifications. All steps were performed on ice or at 4°C with pre-cooled buffers and equipment. Frozen brain tissues were cut into small pieces and crushed using a mortar and pestle, with the base submerged in liquid nitrogen. Liquid nitrogen was placed in the mortar bowl along with the frozen tissue. The pestle was cooled with liquid nitrogen. Crushed tissues were transferred in a clean Dounce homogenizer and homogenized with 50–60 strokes in 1X pre-extraction buffer containing DTT and protease inhibitor cocktail. Tissue lysates were incubated on ice for 15 min and centrifuged for 10 min at 12,000 rpm at 4°C. The supernatant was removed, and pellets were washed with ice cold 1X PBS to avoid cytoplasmic contamination. Nuclear pellets were suspended in extraction buffer with DTT and protease inhibitor and incubated on ice for 15 min, vortexing for 5 s every 3 min. Samples were sonicated briefly and centrifuged for 10 min at 14,000 rpm at 4°C. The supernatant was transferred to clean and cold centrifuge tubes. Extraction cleaner was added to supernatants and kept at RT. After a 20-min incubation, samples were centrifuged for 1 min at 14,000 rpm at 4°C. Supernatants were collected in clean and cold microcentrifuge tubes. Protein estimation was done by Bradford method using Pierce 660 nm Protein Assay Reagent (cat# 22660, ThermoFisher Scientific). Samples were aliquoted and stored at −80°C until further use.
HDAC activity assay
HDAC activity/inhibition assay was done utilizing the Epigenase HDAC Activity/Inhibition Direct Assay Kit (Colorimetric; Cat# P-4034, Epigentek) per the manufacturer's instructions. Standards and samples were diluted with HDAC assay buffer. Each HDAC assay standard (1 µl) was added to 49 µl of HDAC assay buffer, totaling 50 µl per well. Blank wells included 49 μl HDAC assay buffer with 1 µl of HDAC substrate, totaling 50 µl. For samples, 10 µg of each nuclear extract, prepared as described above, was added to 1 µl of HDAC substrate (stock 50 µg/ml) and HDAC assay buffer to a final volume of 50 μl per well. As a positive control for inhibition, 5 µl of HDACi trichostatin A (stock 100 μM, provided with the kit) was added to 10 µg of extract from untreated controls, 1 µl HDAC substrate, and HDAC assay buffer to a final volume of 50 µl. Samples were analyzed in duplicate. Strip-well microplates were tightly covered with adhesive film to avoid evaporation and were incubated at 37°C for 90 min. The reaction solution from each well was removed, and each well was washed three times with 150 μl of the diluted 1X wash buffer. For antibody binding and signal enhancing, 50 μl of the diluted capture antibody was added to each well, then covered with aluminum foil and incubated for 60 min at RT. Capture antibody was removed from each well and washed, as above. The diluted detection antibody (50 μl) was added to each well, and the plate was covered and incubated for 30 min at RT. Detection antibody solution from each well was removed and each well was washed as before. Detection solution (100 μl) was added to each well and the plate was incubated at RT for 10 min, away from light. When the color in the positive control well medium turned blue,100 μl of stop solution was added to each well to stop the enzyme reaction. Absorbance was read on a SpectraMax PLUS microplate reader (Molecular Devices, San Jose, CA, USA), within 10 min at 450 nm with reference wavelength of 655 nm. HDAC activity was calculated using the equation:
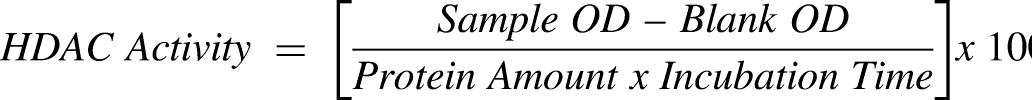
Measurement of Aβ40 and Aβ42 by ELISA
Brain lysates were analyzed for Aβ40 (aa1–40) and Aβ42 (aa1–42) using commercially available kits (Quantikine ELISA kits for human Aβ40, cat#DAB140B, and human Aβ42, cat#DAB142, R&D Systems, Minneapolis, MN, USA), following the manufacturer's instructions. In brief, tissues were rinsed and homogenized with cold 1XPBS. An equal volume of RIPA buffer containing protease inhibitors was added, and tissues were lysed on ice for 30 min with gentle agitation. Samples were centrifuged for 10 min and supernatants were used to assay Aβ (aa1–40 and 1–42). Supernatants were diluted with diluent buffer. Samples and standards (200 µl) were added to wells and incubated at 4°C for 2 h. Each well was washed with 400 µl of wash buffer 3 times for Aβ40 and 4 times for Aβ42. Cold human Aβ in 200 µl volume was added to each well and plates were incubated for 2 h at 4°C. After washing the plates as above, 200 µl of substrate solution was added to each well and plates were incubated for 30 min at room temperature, protected from light. The reaction was stopped by adding 50 µl of stop solution to each well and mixing thoroughly. Optical density was measured at 450 nm (OD450) and 540 nm (OD540). OD540 was subtracted from OD450 for background correction. The final concentration of Aβ (pg/mg protein) was calculated by quantifying against a standard curve and multiplying the concentrations by the dilution factor.
Measurement of H2O2 production
H2O2 production was measured using a Peroxide Assay Kit (Cat# ab272537, Abcam) as directed by the manufacturer. In brief, tissue lysates were deproteinized to remove enzymes from samples before the assay. A mixture of probe and detection reagent was added to tissue lysate containing 50 µg of total protein, and the reaction mixture was incubated at RT for 30 min. Samples were analyzed with a SpectraMax PLUS microplate reader (Molecular Devices) at an absorbance of 580 nm. Peroxide content in the samples was determined from the standard curve and expressed in nmol/mg of total protein as described previously. 54
Measurement of total antioxidant capacity (TAC)
Total antioxidant capacity was quantified using Total Antioxidant Capacity Assay Kit (Cat# ab65329, Abcam) per the manufacturer's protocol. The method is based on the reduction of Cu2+ to Cu+ by antioxidants and other reducing equivalents in the sample. Cu+ interacts with a chromophore to produce a colored product with an absorption maximum at 570 nm. Briefly, brain tissue samples were homogenized in ice-cold phosphate-buffered saline (pH 7.4) with a Dounce homogenizer with 10–15 strokes. Tissue homogenates were incubated for 10 min on ice. Samples were centrifuged in microcentrifuge tubes at 4°C at 12,000 RPM for 5 min. Supernatants were collected and used for the assay. Each sample or standard in duplicate (100 µl) was added to 96-well ELISA plates. Cu2+ working solution (100 µl; prepared by mixing one part Cu2+ with 49 parts of assay diluent) was added to each well and incubated at room temperature for 90 min on an orbital shaker, protected from light. Absorbance was measured by a SpectraMax PLUS microplate reader (Molecular Devices) at 575 nm, with samples and standards corrected against the blank. The net absorbance values of antioxidants were compared with a known Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) standard curve. Results are given as nmol Trolox equivalent antioxidant capacity (TEAC) per mg of tissue protein.
Measurement of 4-HNE protein adducts
A reactive byproduct of lipid peroxidation and a biomarker of OS, 4-HNE was measured via 4-HNE-protein adducts present in brain tissue lysates from VOR-treated and control mice, using a Lipid Peroxidation (4-HNE) Assay Kit (Cat# ab238538, Abcam) according to the manufacturer's instructions. Briefly, 50 µg of tissue lysate was used to measure the adducts. 4-HNE-protein adducts present in the sample or standard were probed with the primary 4-HNE antibody, followed by horseradish peroxidase (HRP)-conjugated secondary antibody. The 4-HNE-protein adduct content in the unknown samples was determined by comparing with a standard curve that was prepared from predetermined 4-HNE-BSA standards as described previously. 55
Measurement of citrate synthase activity
Brain tissue homogenate supernatant samples were prepared from VOR-treated and untreated mice. Citrate synthase (CS) activity was detected using the CS assay kit (Cat# MBP3-24481, Novus Biologicals) following the manufacturer's protocol. Brain tissue homogenates were prepared in extraction buffer using a Dounce homogenizer at 4°C. Homogenates were centrifuged at 1000xg for 15 min. Supernatants were collected, and protein concentrations were estimated. For the assay, 125 µl of buffer solution, 30 µl of the substrate, and 20 µl of the chromogenic agent were added to each well and mixed thoroughly for 3 s using the shaker in the microplate reader. OD values were measured at 412 nm (absorbance reading A1) using the microplate reader. Samples (plate) were then incubated at 37°C for 8 min, mixed thoroughly for 3 s, and measured again at 412 nm as absorbance reading A2. This allowed the calculation of the ΔA412 value, defined as A2 minus A1. A standard curve was plotted with ΔA412 values. CS enzyme activity was calculated as:
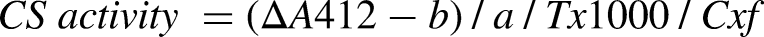
Measurement of ATP levels
ATP levels were measured in deproteinized tissue samples using an ATP Assay Kit (Colorimetric/Fluorometric; Cat# ab83355, Abcam) according to the manufacturer's instructions. Reaction mix containing ATP reaction buffer, ATP probe, ATP converter, and developer mix was added to tissue lysates. The reaction mixture was incubated at RT for 30 min. The absorbance was measured at 570 nm. The concentration of ATP in the samples was determined using a standard curve of known ATP concentrations.
Statistical analysis
All analyses were performed using Prism 10.2.1 (GraphPad, San Diego, CA). Results are graphed as bar graphs with SEM or violin plots with medians. Descriptive statistics in the results text indicate mean ± SD. For each measure, normality was tested within each of the 6 groups (3 treatments×2 sexes) using the Shapiro-Wilk test. Most measures showed normal distributions except for AH3 and H3 expression, 4-HNE adducts, CS activity, and H2O2, each of which showed a minor deviation from normality (p = 0.0214 to p = 0.0485) in only 1/6 groups. Therefore, we employed ANOVAs, which are robust against minor deviations from normality. The effect of diet on AH3 and H3 was analyzed using one-way ANOVA with Dunnett's multiple comparisons test. In the remaining assays, comparisons between sexes were possible, and differences between treatments, sexes or time points in the 14-day diet test were analyzed using two-way AVOVA with Dunnett's multiple comparisons tests. Correlation analyses used Spearman's r, followed by simple linear regression to further examine correlations of interest. The goodness of fit was compared between males and females by comparing simple linear regression R squared (R2) values between sexes using a Shapiro-Wilk normality test followed by a two-tailed t-test with Welch's correction. For the extended 9-week test of the 0.18 mg/g VOR diet, repeated measures one-way ANOVA with Dunnet's multiple comparisons test was used to examine change in weight across time points, and two-way repeated measures ANOVA with Sidak's multiple comparisons test was used to compare weight change between the treatment and control group at each time point. p values of <0.05 were considered statistically significant.
Results
A low dose of dietary VOR was tolerable for 14 days
To examine VOR administration via diet, we conducted an experiment in which VOR was administered via pelleted purified diets (0.0 mg/g, 0.18 mg/g, or 0.36 mg/g) over the course of 14 days (D0-D14, Figure 1(a)). hAβ-KI mice were weighed at D0, then moved onto VOR-containing diets. Mouse weights and diet consumption were measured at D7 and D14. Based on measurements of mouse weights and diet consumption over D0-D14, the calculated VOR dose delivered to mice on 0.18 mg/kg VOR diet was 123 mg/kg/week for males, 137 mg/kg/week for females, and 130 mg/kg/week across all mice, achieving 98.6%, 109.5%, and 103.7%, respectively, of the target dose of 125 mg/kg/week. On the 0.36 mg/kg VOR diet, VOR delivered was 240 mg/kg/week for males, 289 mg/kg/week for females, and 262 mg/kg across all mice, achieving 96.0%, 115.8%, and 104.9%, respectively, of the target dose of 250 mg/kg/week.
Since differences in body weight between males and females are known, statistical comparisons of weights did not include sex differences. To address tolerability, we compared body weight across three (D0, D7, and D14) time points separately for both males (Figure 1(b)) and females (Figure 1(c)).
For males, there was a main effect of time (F(1,17) = 51.98, p < 0.0001). A significant interaction between treatment group and time was also noted (F(4,24) = 4.928, p = 0.0048). However, there was no main effect of treatment group (F(2,12) = 0.1277, p = 0.8813). The control (0.0 mg/g) group significantly increased in weight between D0 (36.3 ± 3.2 g) and D7 (38.1 ± 3.2 g, p = 0.0007) and between D0 and D14 (39.4 ± 3.8 g, p = 0.0113, Figure 1(b)). Similarly, the 0.18 mg/g VOR diet group increased in weight between D0 (36.4 ± 1.7 g) and D7 (38.1 ± 2.1 g, p = 0.0283) and also between D0 and D14 (39.0 ± 2.2 g, p = 0.0103). However, for the 0.36 mg/g VOR diet group, a significant difference was observed only between D0 (37.9 ± 2.7 g) and D7 (39.3 ± 2.7 g, p = 0.0087) but not between D0 and D14 (38.8 ± 2.5 g, p = 0.1255). Also, males showed no significant differences in weight on VOR diets compared to control diet at D0 or at later time points (Figure 1(b)).
For females (Figure 1(c)), there was also a main effect of time (F(1,19) = 6.008, p = 0.0128), but no main effect of treatment (F(2,12) = 0.3571, p = 0.7069) or time-treatment interaction (F(4,24) = 0.6767, p = 0.6147). Multiple comparison testing indicated no significant effects within or across treatment groups (p > 0.05).
Finally, we examined percent change (ΔW) in body weight (D14/D7, Figure 1(d)). There was a main effect of treatment (F(2,24) = 5.443, p = 0.0112) but no effect of sex (F(1,24) = 1.379, p = 0.2518) or treatment-sex interaction (F(2,24) = 0.3633, p = 0.6992). ΔW increased in both control (4.0 ± 2.4%) and 0.18 mg/g VOR groups (2.6 ± 2.8%) and did not differ significantly between the two (p = 0.4438). However, compared to the control group, ΔW did not increase in the 0.36 mg/g VOR group (−0.2 ± 3.3%, p = 0.0065). Sex-specific changes in body weight showed significantly reduced weight gain in males on 0.36 mg/g VOR (−1.2 ± 1.4%, p = 0.0436) relative to the control group (3.2 ± 2.7%), but this was not observed in females on 0.36 mg/g VOR (0.8 ± 4.5%, p = 0.0715) compared to controls (4.8 ± 1.9%).
As an additional measure of tolerability, we calculated average daily consumption by weighing diet at D7 and D14 (see Methods; Figure 1(e) and (f)). Among males (Figure 1(e)), there was a main effect of time (F(1,12) = 183.2, p < 0.0001), signifying a small but consistent decrease in consumption between D7 and D14 across all treatment groups. There was no main effect of treatment (F(2,12) = 0.1001, p = 0.9055), but there was a significant time-treatment interaction (F(2,12) = 11.99, p = 0.0014). On control diet, males consumed less at D14 (3.6 ± 0.3) than at D7 (3.9 ± 0.3 g, p = 0.0003; Figure 1(e)). Similar reductions were observed for the 0.18 mg/g VOR group (D7:3.9 ± 0.4 g; D14:3.5 ± 0.3 g, p < 0.0001) and 0.36 mg/g VOR group (D7:4.0 ± 0.3 g; D14: 3.4 ± 0.2 g, p < 0.0001).
Among females (Figure 1(f)), there was a main effect of time (F(1,12) = 17.39, p = 0.0013), but no main effect of treatment (F(2,12) = 3.683, p = 0.0566) or time-treatment interaction (F(2,12) = 3.855, p = 0.0509). On the control diet, females consumed similar amounts between D0−7 (3.2 ± 0.2 g) and D8−14 (3.1 ± 0.2 g, p = 0.8903). However, similar to males, females on the 0.18 mg/g VOR group (D7: 3.9 ± 0.5 g; D14: 3.4 ± 0.3 g, p = 0.0038) and 0.36 mg/g VOR diet (D7:3.9 ± 0.5 g; D14:3.4 ± 0.4 g, p = 0.0043) consumed less food.
Finally, we examined the percent change in diet consumption (ΔC), comparing D7 to D14 (Figure 1(g)). There was a main effect of treatment (F(2,24) = 8.742, p = 0.0014). However, there was neither a main effect of sex (F(1,24) = 1.938, p = 0.1766) nor a treatment-sex interaction (F(2,24) = 1.799, p = 0.1870). Among males, ΔC did not differ between the control (−6.8 ± 3.4%) and 0.18 mg/g (−9.3 ± 2.8%, p = 0.6898) groups. However, compared to the control group, ΔC was less in the 0.36 mg/g (−15.7 ± 1.0%, p = 0.0288) group. Among females, ΔC was less in the 0.18 mg/g group (−11.7 ± 6.5%) than control group (−0.5 ± 3.8%, p = 0.0060) and less in the 0.36 mg/g group (−11.3% ± 9.8%, p = 0.0080) than control group.
Taken together, the 0.18 mg/g group sustained increases in ΔW that were comparable to the control group, particularly among males (Figure 1(d)). Moreover, ΔC was similar between control and 0.18 mg/g groups, particularly among males. However, the 0.36 mg/g VOR diet, particularly among males, was associated with a flattened increase in weight (Figure 1(d)). Overall, these results suggest that 0.18 mg/g is more tolerable than the 0.36 mg/g diet.
Dietary VOR increased brain acetyl-histone H3 (AH3) levels
To determine the extent that VOR-containing diet inhibits HDACs and increases brain histone acetylation in vivo, we compared brain AH3 expression levels and HDAC enzyme activity between treatment groups (Figure 2). WB results for AH3 and H3 expression are shown from total brain tissue lysates from male (Figure 2(a) and (b)) and female (Figure 2(c) and (d)) mice. Relative AH3 and H3 expression were quantified using densitometry normalized to β-actin as a loading control (Figure 2(e) and (f)). In males, there was a significant main effect of treatment (F(2,12) = 7.250, p = 0.0086) on AH3 expression. Multiple comparisons revealed that relative to the control group (1.00 ± 0.16), there were significant increases in AH3 expression in the 0.18 mg/g (1.67 ± 0.44, p = 0.0060) and 0.36 mg/g (1.50 ± 0.16, p = 0.0341) VOR groups. In females, there was a significant main effect of treatment on AH3 (F(2,12) = 3.968, p = 0.0475). Relative to the control group (1.00 ± 0.55), AH3 expression was higher in the 0.18 mg/g group (2.92 ± 1.73, p = 0.0477) but not the 0.36 mg/g VOR (2.78 ± 1.02, p = 0.0667) group.
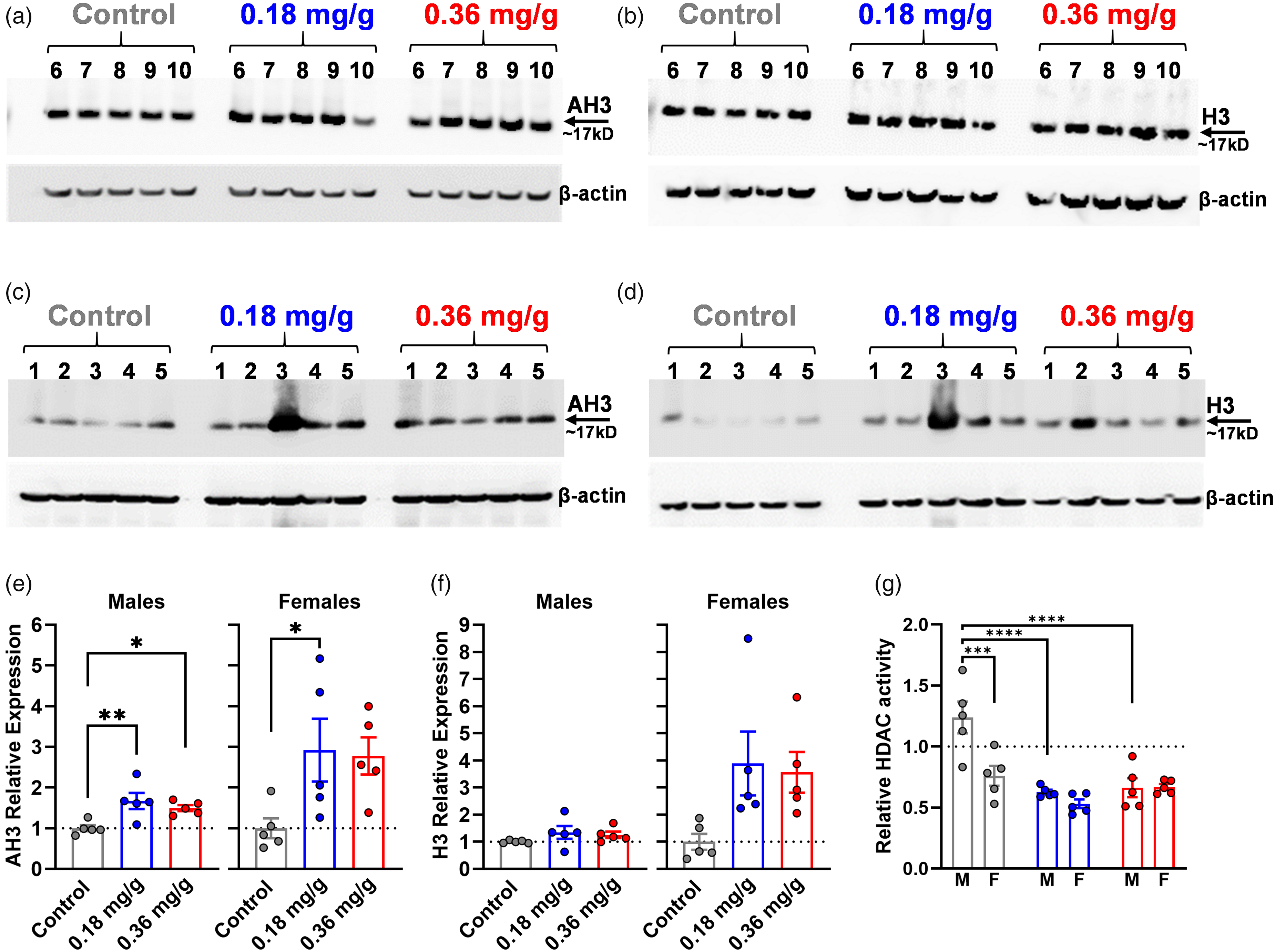
Dietary VOR increased brain acetyl-histone H3 (AH3) levels and reduced brain HDAC activity. Western blots of acetyl-histone H3 (AH3) and total histone H3 (right) expression are shown for (a, b) males or (c, d) females. (e–g) Treatment groups correspond to colors in a-d; M: males, F: females. Densitometry analysis of (e) AH3 expression and (f) total histone H3, normalized to β-actin, are shown. (g) Brain tissue HDAC enzymatic activity across treatment and sex. The relative enzymatic activity of all samples was normalized to the average of control mice, which was defined equal to 1 (dotted line). Statistical significance is denoted by asterisks: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
In contrast to AH3, males showed no significant main effect of diet on H3 expression (F(2,12) = 1.367, p = 0.2918). Relative to the control group (1.00 ± 0.05), the change in total H3 expression of the 0.18 mg/g (1.35 ± 0.53, p = 0.2319) and 0.36 mg/g (1.26 ± 0.26, p = 0.4138) VOR groups did not reach significance. In females, there was no significant main effect of diet (F(2,12) = 3.689, p = 0.0564). Relative to controls (1.00 ± 0.66), total H3 expression did not change significantly in 0.18 mg/g (3.89 ± 2.63, p = 0.0520) and 0.36 m/g (3.56 ± 1.68, p = 0.0855) VOR groups, although a trend toward increased H3 with VOR was observed.
Finally, we examined HDAC enzymatic activity (Figure 2(g)). There were significant main effects of treatment (F(2,24) = 18.51, p < 0.0001) and sex (F(1,24) = 10.16, p = 0.0040) as well as a treatment-sex interaction (F(2,24) = 6.119, p = 0.0071). Relative to the control group (1.00 ± 0.34), HDAC activity was reduced for both the 0.18 mg/g (0.58 ± 0.08, p < 0.0001) and 0.36 mg/g (0.67 ± 0.12, p = 0.0002) VOR groups. In males, compared to the control group (1.24 ± 0.29), HDAC activity was significantly reduced in the 0.18 mg/g (0.63 ± 0.04, p < 0.0001) and 0.36 mg/g groups (0.66 ± 0.17, p < 0.0001). In females, compared to the control group (0.76 ± 0.18), HDAC activity in 0.18 mg/g (0.53 ± 0.08, p = 0.0649) and 0.36 mg/g (0.67 ± 0.05, p = 0.5851) VOR groups did not differ significantly. Interestingly, the largest sex-specific difference was in the control group; males (1.24 ± 0.29) had higher basal HDAC activity compared to females (0.76 ± 0.18, p = 0.0001), but this difference was normalized in the 0.18 mg/g (p = 0.3574) and 0.36 mg/g (p = 0.9557) VOR groups.
Dietary VOR did not significantly impact Aβ levels in hAβ-KI mice
Although the original study of the hAβ-KI model did not find evidence of plaque formation, soluble and insoluble Aβ40 and Aβ42 were detected. 42 Soluble forms of amyloid β (Aβ) may be pathogenically important in AD and thus have both diagnostic and therapeutic importance. 56 To examine the extent to which VOR affects Aβ in hAβ-KI mice, we measured Aβ40 (aa1–40) and Aβ42 (aa1–42) in the brain tissue lysates of control and VOR treated mice.
Aβ40 (Figure 3(a)) exhibited a significant main effect of sex (F(1,24) = 4.765, p = 0.0391), but not treatment (F(2,24) = 1.588, p = 0.2250), and no treatment-sex interaction (F(2,24) = 0.3438, p = 0.7125). Relative to controls (1764 ± 459 pg Aβ40/mg protein), there were trends toward reduction that were not statistically significant for the 0.18 mg/g VOR (1443 ± 432 pg/mg, p = 0.1542) and 0.36 mg/g VOR groups (1582 ± 368 pg/mg, p = 0.5072). Males showed no significant change with 0.18 mg/g (1230 ± 444 pg/mg, p = 0.3221) or 0.36 mg/g (1507 ± 413 pg/mg, p = 0.9558) VOR relative to controls (1570 ± 331 pg/mg). Similarly, females showed no significant change with 0.18 mg/g (1657 ± 328 pg/mg, p = 0.4054) or 0.36 mg/g (1657 ± 347 pg/mg, p = 0.4062) relative to controls (1957 ± 521 pg/mg). Despite the significant main effect of sex, there were no significant within-group sex differences in Aβ40 for any diet (p > 0.05).
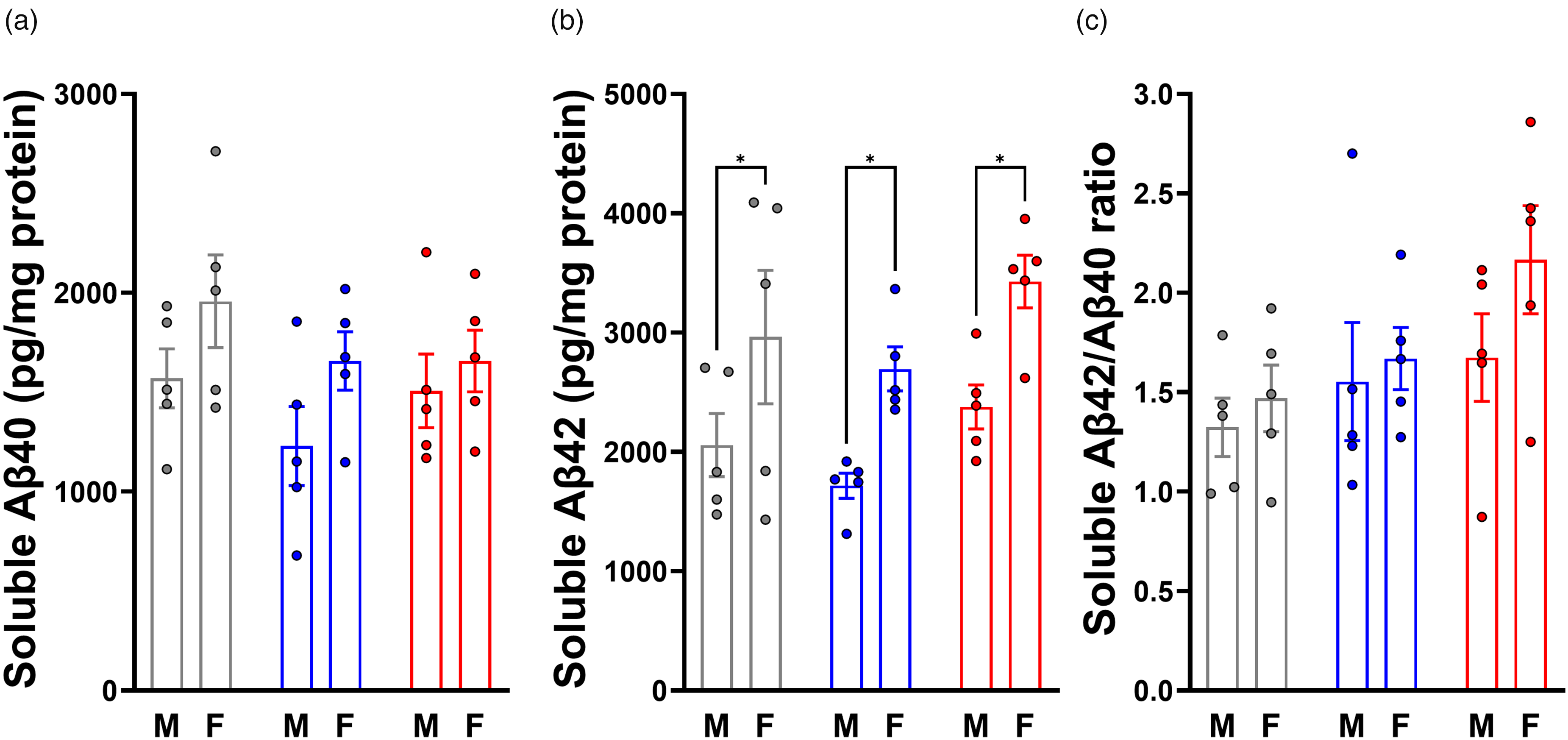
Soluble Aβ40, Aβ42, and Aβ42/Aβ40 ratio is affected by sex but not VOR treatment. (a) Soluble Aβ40 and (b) soluble Aβ42 were measured by ELISA, and the Aβ42/Aβ40 ratio was calculated (c).
Similarly to Aβ40 level, Aβ42 level (Figure 3(b)) exhibited a significant main effect of sex (F(1,24) = 16.93, p = 0.0004), but not treatment (F(2,24) = 2.878, p = 0.0758), and no treatment-sex interaction (F(2,24) = 0.03079, p = 0.9697). Relative to Aβ42 levels in controls (2510 ± 1038 pg/mg), mice on 0.18 mg/g VOR showed no significant difference (2206 ± 605 pg/mg, p = 0.4852), and mice on 0.36 mg/g VOR also showed no significant difference (2903 ± 700, p = 0.3151). The same general pattern in Aβ42 level was seen when males and females were examined independently. Relative to males on control diet (2057 ± 591 pg/mg), males on 0.18 mg/g VOR diet Aβ42 levels were not significant (1717 ± 235 pg/mg, p = 0.6261). Males on 0.36 mg/g VOR diet also showed no significant change in Aβ42 levels (2378 ± 411 pg/mg, p = 0.6571). Similarly, relative to females on control diet (2963 ± 1249 pg/mg), the 0.18 mg/g VOR group did not show a significant difference in Aβ42 levels (2695 ± 411 pg/mg, p = 0.7436), while those in the 0.36 mg/g VOR group also showed no significant difference (3428 ± 492, p = 0.4325). Females expressed significantly elevated Aβ42 relative to males on control diet (p = 0.0377), 0.18 mg/g VOR diet (p = 0.0258), as well as 0.36 mg/g VOR diet (p = 0.0176).
Finally, we calculated the Aβ42/Aβ40 ratio (Aβ42/40) for each mouse (Figure 3(c)). We did not observe significant main effects of diet (F(2,24) = 2.927, p = 0.0729) or sex (F(1,24) = 1.998, p = 0.1703) on Aβ42/40, and there was no treatment-sex interaction (F(2,24) = 0.4612, p = 0.6360). Despite the lack of a main effect of diet, multiple comparisons testing found that Aβ42/40 significantly increased in the 0.36 mg/g VOR group (1.92 ± 0.58, p = 0.0443) compared to the control group (1.40 ± 0.34). There was no significant difference in Aβ42/40 between the control and 0.18 mg/g groups (1.61 ± 0.50, p = 0.5209). There were no significant sex-specific differences in Aβ42/40 between males on control diet (1.32 ± 0.33) and those on 0.18 mg/g (1.55 ± 0.66, p = 0.6802) or 0.36 mg/g (1.68 ± 0.49, p = 0.4275) VOR diets. Similarly, there were no differences in Aβ42/40 between the female control group (1.47 ± 0.37) and the 0.18 mg/g (1.67 ± 0.35, p = 0.7432) or 0.36 mg/g (2.17 ± 0.61, p = 0.0597) VOR groups. Finally, multiple comparisons testing found no significant differences in Aβ42/40 between males and females of any individual treatment group (p > 0.05). Overall, our results demonstrate that the effect of two weeks of VOR on levels of Aβ40 and Aβ42 was minor.
Sex-specific effects on synaptic proteins were independent of VOR treatment
Synaptic degeneration occurs in AD. 57 As an additional measure of AD pathology, we used WB to compare postsynaptic density protein-95 (PSD-95, Figure 4(a) and (b)) and the presynaptic marker synaptophysin (syn, Figure 4(c) and (d)) between treatment groups. For PSD-95 (Figure 4(b)), a significant main effect of sex was observed (F(1,23) = 6.124, p = 0.0211), but there was no main effect of treatment (F(2,23) = 2.897, p = 0.0755) and no treatment-sex interaction (F(2,23) = 1.192, p = 0.3218). Relative to mice on control diet (0.99 ± 0.22), PSD-95 expression was not significantly different in those fed 0.18 mg/g VOR (1.07 ± 0.16, p = 0.5299) or 0.36 mg/g VOR diets (0.91 ± 0.10, p = 0.3091). Among males, there was no significant difference between those on 0.18 mg/g VOR (1.13 ± 0.21, p = 0.9999) or 0.36 mg/g VOR diets (0.93 ± 0.14, p = 0.1033) and those on control diet (1.13 ± 0.23). Similarly, PSD-95 expression did not differ significantly between females on 0.18 mg/g VOR (1.01 ± 0.08, p = 0.2828) or 0.36 mg/g VOR diets (0.88 ± 0.03, p = 0.9886) and those on control diet (0.87 ± 0.14). Within control diets, males showed significantly higher PSD-95 expression than females (p = 0.0189); however, no significant sex differences were observed on 0.18 mg/g (p = 0.2254) or 0.36 mg/g VOR diets (p = 0.6575).
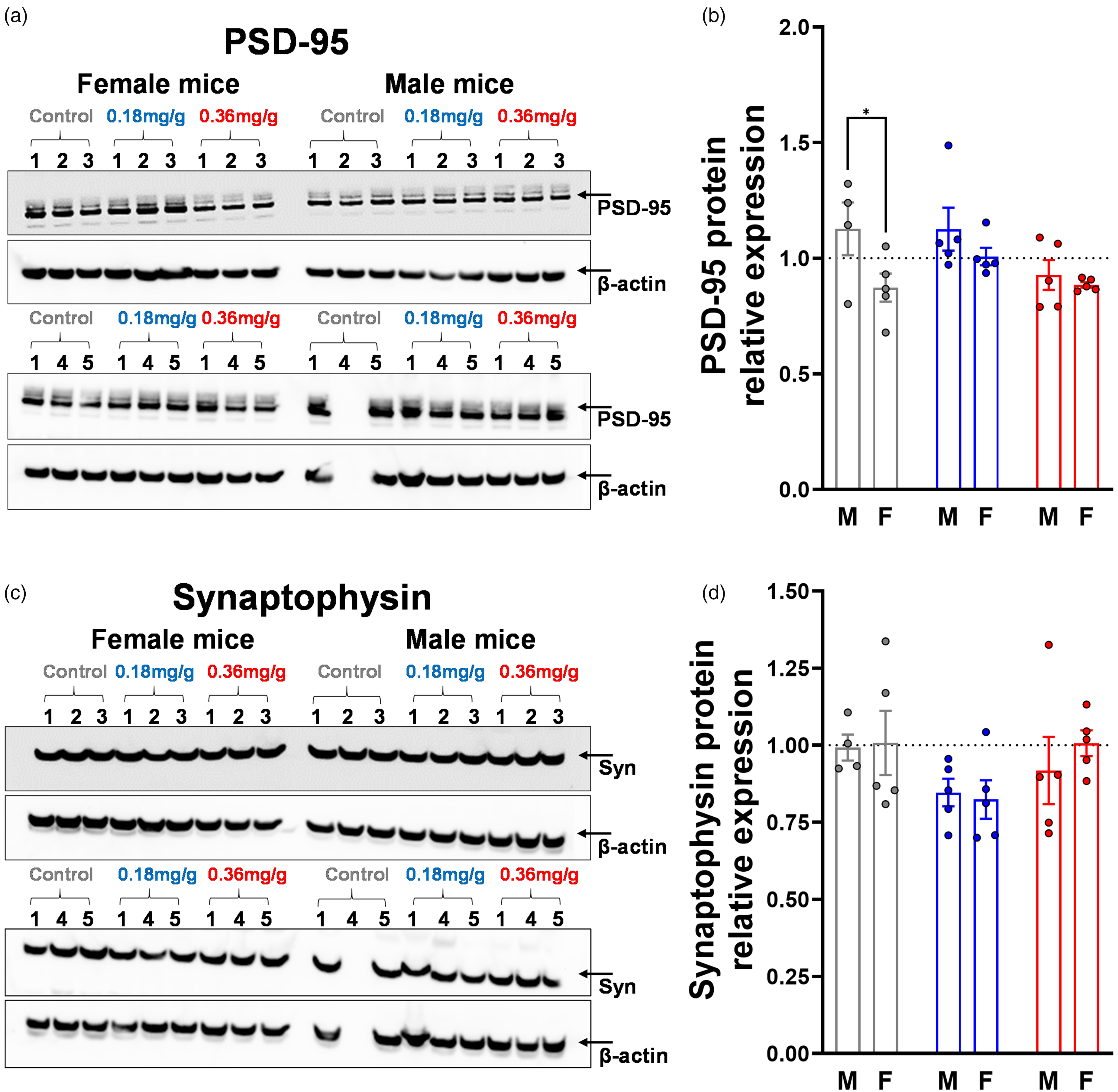
Synaptic proteins PSD-95 and synaptophysin were not significantly affected by dietary VOR. (a) Western blot of PSD-95 with (b) graph of normalized expression level. (c) Western blot of synaptophysin with (d) graph of normalized expression level. The average of males and females was defined as equal to 1 (dotted lines). β-actin was used as a loading control. All groups numbered n = 5 mice, except for males on control diet which numbered n = 4 due to the lack of protein in one sample. *p < 0.05. Samples for PSD-95 and synaptophysin were run across two western blots each, with the first sample from each treatment-sex group (6 in total) included on both blots as calibrators to allow plotting on one graph. The averages of the synaptophysin:β-actin or PSD-95:β-actin ratios of the six repeated samples were used to re-scale blot 2 to match blot 1.
Syn (Figure 4(d)) exhibited no significant main effects of treatment (F(2,23) = 2.621, p = 0.0943) or sex (F(1,23) = 0.1929, p = 0.6646) and no treatment-sex interaction (F(2,23) = 0.2901, p = 0.7509). There were no significant differences in relative syn expression between mice on control diet (1.00 ± 0.17) and those on 0.18 mg/g VOR (0.84 ± 0.12, p = 0.0743) or 0.36 mg/g VOR diets (0.96 ± 0.18, p = 0.8376). Multiple comparisons testing for sex-specific effects revealed no significant differences between the male control group (0.99 ± 0.08) and the0.18 mg/g VOR (0.85 ± 0.10, p = 0.3275) or 0.36 mg/g VOR groups (0.92 ± 0.24, p = 0.7230). Similarly, there were no significant differences between the female control group (1.01 ± 0.23) and the 0.18 mg/g VOR (0.82 ± 0.14, p = 0.1619) or 0.36 mg/g VOR groups (1.01 ± 0.09, p > 0.9999). Finally, no significant differences in syn expression were observed between males and females on any diet (all p > 0.05).
Low-dose dietary VOR reduced measures of oxidative stress
We examined the levels of H2O2, total antioxidant capacity (TAC), and 4-HNE protein adducts in brain tissue lysates from control and VOR-treated mice. For H2O2 level (Figure 5(a)), there were main effects of treatment (F(2,24) = 15.34, p < 0.0001) and sex (F(1,24) = 10.35, p = 0.0037), but there was no treatment-sex interaction (F(2,24) = 2.935, p = 0.0724). Compared to the control group (7.70 ± 1.33 nmol/mg), H2O2 level was significantly reduced in 0.18 mg/g (4.47 ± 1.99 nmol/mg, p < 0.0001) and 0.36 mg/g (5.46 ± 1.50 nmol/mg p = 0.0019) VOR groups. For males, relative to control diet (7.67 ± 1.85 nmol/mg), H2O2 level was significantly reduced in 0.18 mg/g (3.00 ± 1.04 nmol/mg, p < 0.0001) and 0.36 mg/g (4.61 ± 1.51 nmol/mg, p = 0.0026) groups. However, within females, compared to the control group (7.73 ± 0.75 nmol/mg), H2O2 level was similar in the 0.18 mg/g VOR (5.94 ± 1.56 nmol/mg, p = 0.0807) and 0.36 mg/g VOR (6.32 ± 0.96 nmol/mg, p = 0.1865) groups. On the 0.18 mg/g VOR diet, males were associated with significantly lower H2O2 level than females (p = 0.0019), but H2O2 levels were similar between sexes in control (p = 0.9477) and 0.36 mg/g (p = 0.0541) groups.
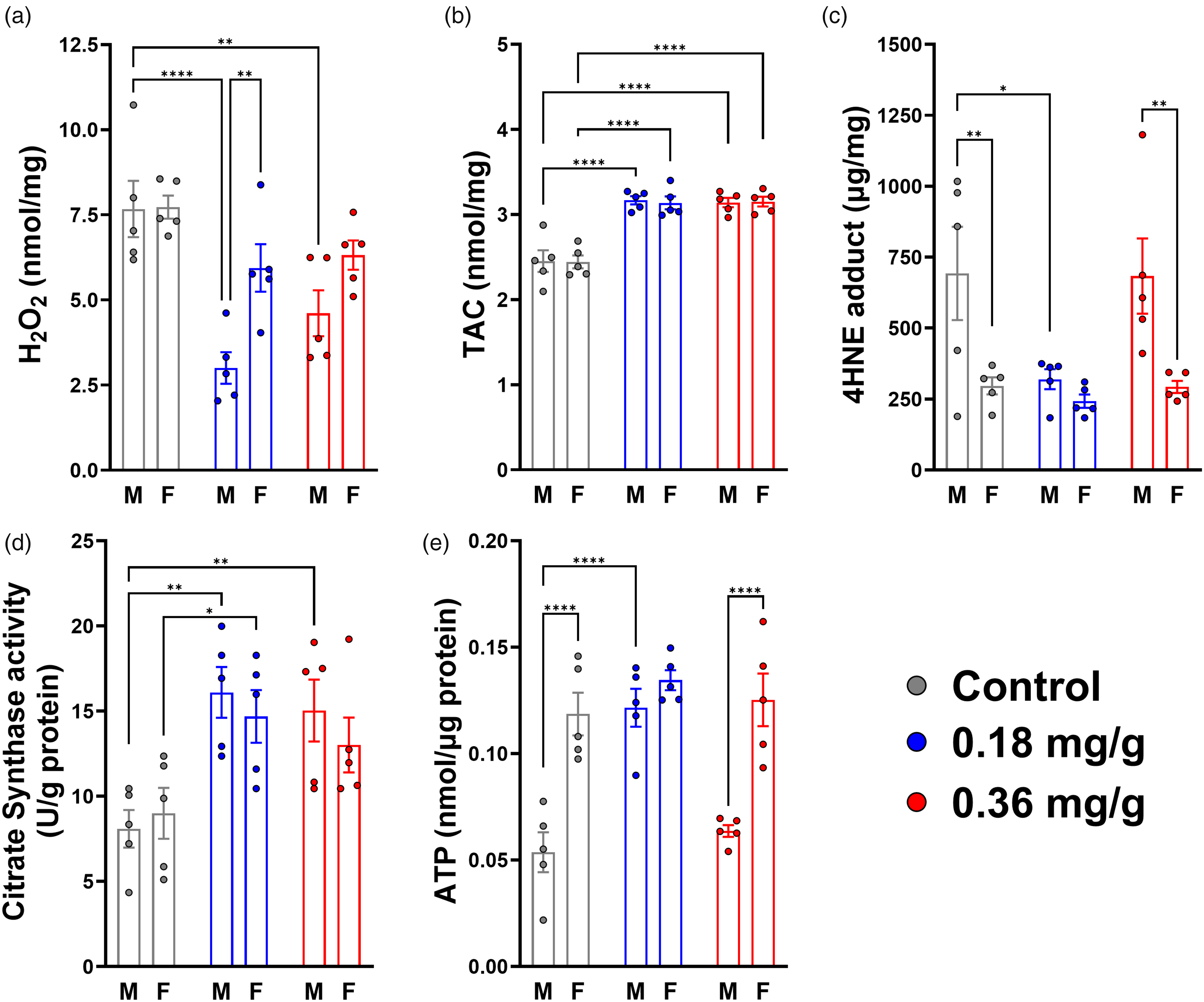
Low-dose dietary VOR reduced measures of OS and improved mitochondrial health. (a) H2O2, (b) total antioxidant capacity (TAC), (c) lipid peroxidation (4-hydroxynonenal, or 4-HNE, adduct), (d) citrate synthase activity and (e) ATP level. Colors indicate treatment groups: control (0.0 mg/g, gray), 0.18 mg/g (blue), and 0.36 mg/g (red) vorinostat. M: male, F: female. Statistical significance is denoted by asterisks: *p < 0.05; **p < 0.01; ****p < 0.0001.
For TAC (Figure 5(b)), the main effect of treatment was significant (F(2,24) = 54.34, p < 0.0001), but not sex (F(1,24) = 0.02163, p = 0.8843) or treatment-sex interaction (F(2,24) = 0.03536, p = 0.9653). Relative to the control group (2.45 ± 0.22 nmol/mg), TAC was higher in 0.18 mg/g (3.15 ± 0.13, p < 0.0001) and 0.36 mg/g VOR (3.15 ± 0.12, p < 0.0001) groups. In males, compared to the control diet (2.45 ± 0.28 nmol/mg), TAC was significantly increased in the 0.18 mg/g (3.17 ± 0.11 nmol/mg, p < 0.0001) and 0.36 mg/g VOR groups (3.14 ± 0.12 nmol/mg, p < 0.0001). Similarly, in females, TAC was increased in the 0.18 mg/g (3.14 ± 0.17 nmol/mg, p < 0.0001) and 0.36 mg/g VOR (3.15 ± 0.13 nmol/mg, p < 0.0001) groups compared to the control group (2.44 ± 0.17 nmol/mg). No significant differences in TAC were observed between sexes (p > 0.05).
For lipid peroxidation, as assessed by 4-HNE adducts (Figure 5(c)), there were main effects of treatment (F(2,24) = 3.700, p = 0.0397) and sex (F(1,24) = 15.64, p = 0.0006), but no treatment-sex interaction (F(2,24) = 2.093, p = 0.1452). Compared to the control group (494.7 ± 325.1 μg/mg), there was a significant reduction in 4-HNE level in the 0.18 mg/g group (281.3 ± 75.3 μg/mg, p = 0.0457), but not in the 0.36 mg/g VOR group (488.2 ± 287.3 μg/mg, p = 0.9961). Similarly, in males, compared to the control group (692.7 ± 367.8 μg/mg), 4-HNE level was reduced in the 0.18 mg/g VOR group (319.9 ± 79.4 μg/mg, p = 0.0130), but was similar between control and 0.36 mg/g VOR groups (683.7 ± 296.3 μg/mg, p = 0.9962). However, in females, the control group (296.6 ± 67.3 μg/mg) was not different from the 0.18 mg/g (242.6 ± 52.0 μg/mg, p = 0.8763) or 0.36 mg/g VOR groups (292.7 ± 47.6 μg/mg, p = 0.9993). Sex-specific differences in 4-HNE level were apparent within the control (p = 0.0045) and 0.36 mg/g VOR groups (p = 0.0049), but levels within the 0.18 mg/g group were comparable (p = 0.5456).
Vorinostat diet improved measures of mitochondrial health
As indicators of mitochondrial function, we measured ATP and citrate synthase (CS) activity levels in brain tissue lysates from hAβ-KI mice fed with control or VOR-containing diets. CS activity (Figure 5(d)) showed a significant main effect of treatment (F(2,24) = 11.27, p = 0.0004). The effect of sex was not significant (F(1,24) = 0.4535, p = 0.5071) and there was no treatment-sex interaction (F(2,24) = 0.5157, p = 0.6036). Multiple comparisons testing revealed significant increases in brain CS activity in mice fed 0.18 mg/g (15.4 ± 3.3 U/g protein, p = 0.0003) or 0.36 mg/g VOR diets (14.0 ± 3.8 U/g, p = 0.0028) relative to controls (8.5 ± 2.8 U/g). A sex-specific analysis revealed significant increases in CS activity in males fed 0.18 mg/g (16.1 ± 3.3 U/g, p = 0.0021) or 0.36 mg/g VOR diet (15.0 ± 4.1 U/g, p = 0.0070) relative to control diet (8.1 ± 2.5 U/g). CS activity in females differed significantly only between control (9.0 ± 3.3 U/g) and the 0.18 mg/g group (14.7 ± 3.5 U/g, p = 0.0270), but not between control and the 0.36 mg/g group (13.0 ± 3.6 U/g, p = 0.1335). There were no differences in CS activity between sexes within any treatment group (p > 0.05).
Measuring ATP levels (Figure 5(e)), there were both main effects of treatment (F(2,24) = 13.08, p < 0.0001) and sex (F(1,24) = 43.05, p < 0.0001), as well as a significant treatment-sex interaction (F(2,24) = 5.622, p = 0.0099). Relative to the control group (0.086 ± 0.040 nmol/μg), ATP level was significantly increased in the 0.18 mg/g VOR group (0.128 ± 0.017 nmol/μg, p = 0.0001). However, ATP level was similar between the control and 0.36 mg/g VOR group (0.094 ± 0.038 nmol/μg, p = 0.5392). Within males, relative to the control group (0.054 ± 0.021 nmol/μg), 0.18 mg/g VOR diet significantly increased brain ATP levels (0.122 ± 0.020 nmol/μg, p < 0.0001). However, compared to the control group, ATP levels were similar in the 0.36 mg/g VOR group (0.064 ± 0.006 nmol/μg, p = 0.6344). In contrast, within females, relative to the control group (0.119 ± 0.022 nmol/μg), ATP levels were not significantly different for the 0.18 mg/g (0.135 ± 0.011 nmol/μg, p = 0.3413) or 0.36 mg/g VOR groups (0.125 ± 0.028 nmol/μg, p = 0.8120). Finally, there were significant sex-specific differences in ATP level between males and females. ATP levels were higher in females in both control (p < 0.0001) and 0.36 mg/g VOR groups (p < 0.0001), but males and females were comparably high in the 0.18 mg/g group (p = 0.3020). These results further corroborated the finding that brains from male mice had higher basal oxidative damage than females (4-HNE, Figure 5(c)). Taken together, our results reveal complex, sex-specific effects of VOR on measures related to OS and mitochondrial health, generally consistent with improved effect at the lower (0.18 mg/g) dose.
HDAC enzyme activity correlates negatively with AH3 expression, TAC, and ATP levels, but correlates positively with H2O2 level
We observed that several independent measures were spread over wide dynamic ranges. Therefore, we created a correlation matrix to graphically visualize the extent to which these measures correlated, independent of treatments (Figure 6(a)). Several significant positive (blue) and negative (red) correlations were present (Figure 6(a)). First, we observed significant correlations between VOR dose and AH3 level (r = 0.60, p = 0.0004) and TAC (r = 0.71, p < 0.0001) as well as moderately significant correlations with weight change (r = −0.44, p = 0.0159), H3 level (r = 0.52, p = 0.0033), HDAC activity (r = −0.39, p = 0.0324), and H2O2 (r = −0.47, p = 0.0085). HDAC enzymatic activity also showed significant correlations with several measures, including AH3 expression (r = −0.64, p = 0.0001), H3 expression (r = −0.55, p = 0.0015), ATP (r = −0.50, p = 0.0048), and TAC (r = −0.59, p = 0.0006). Finally, as would be expected for an HDACi, the directionality of the Spearman's r values for HDAC activity tended to be opposite those observed for VOR dose level.
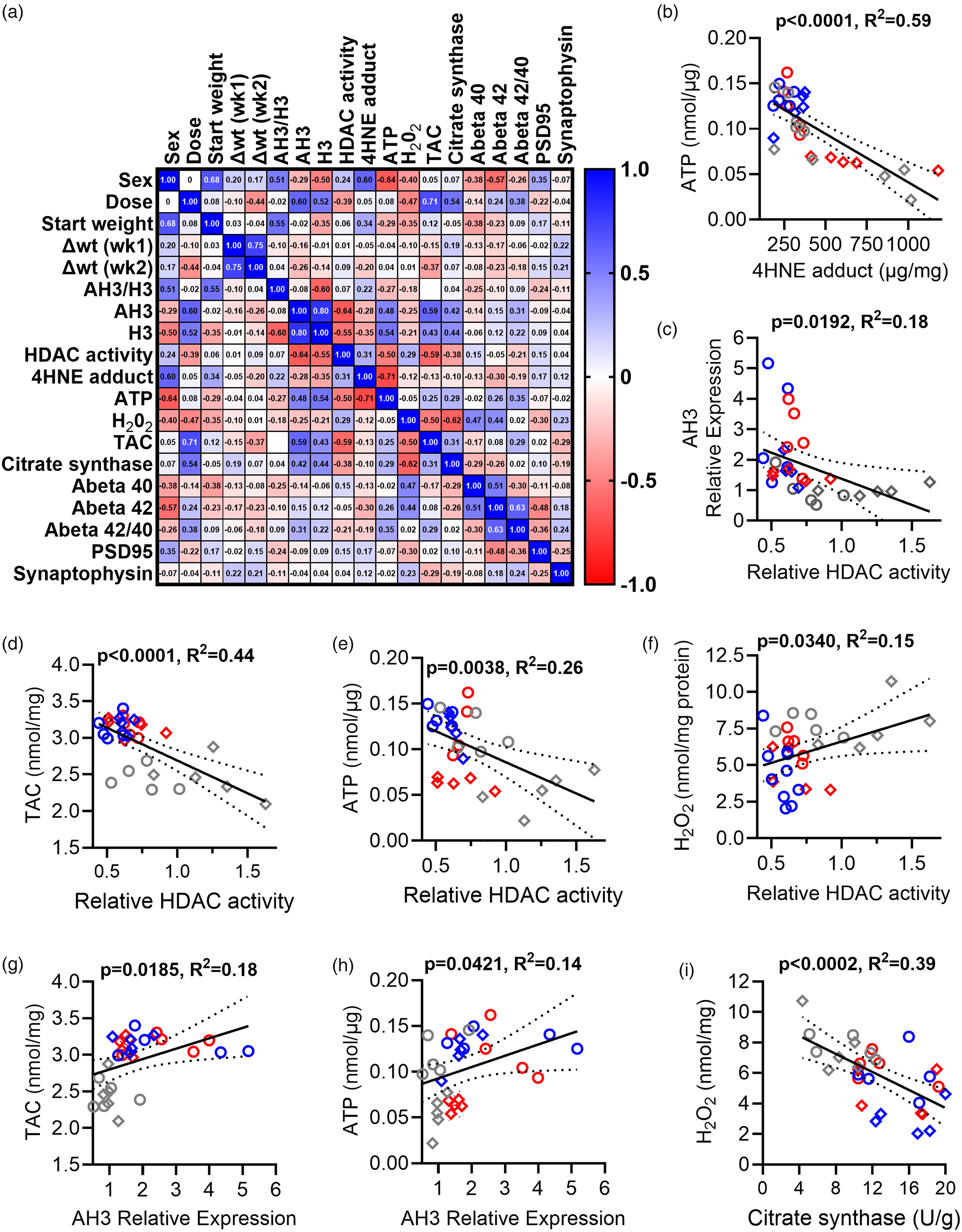
Relationships between HDAC and OS measures. (a) Correlation matrix showing Spearman's r for each comparison. Positive (blue) and negative (red) correlations are shown. (b–h) Pairs of parameters were analyzed using linear regression (solid line; 95% confidence intervals (CI) shown as dotted lines). VOR doses are denoted as 0.0 mg/g (control, grey), 0.18 mg/g (blue), or 0.36 mg/g (red) VOR. Females (open circles) and males (open diamonds) also are indicated. (b) A negative relationship between 4-HNE and ATP was found. (c–f) Relationships between HDAC enzyme activity and AH3 (c), TAC (d), ATP level (e), and H2O2 level (f) are shown. (g and h) Relationships between AH3 expression and TAC (g) or ATP level (h). (i) Relationship between citrate synthase activity and H2O2.
To explore these relationships further, we then investigated select pairs of measures using simple linear regression (Figure 6(b)–(h)). As expected, based on the toxic nature of 4-HNE adduct generation, we found a strong negative relationship between ATP and 4-HNE levels (p < 0.0001, R2 = 0.59, Figure 6(b)). Moreover, as expected, HDAC activity negatively correlated with AH3 expression (p = 0.0192, R2 = 0.18, Figure 6(c)).
We then investigated relationships between HDAC activity and OS measures (TAC, ATP, and H2O2). Linear regression revealed a significant negative relationship between HDAC activity and TAC (p < 0.0001, R2 = 0.44, Figure 6(d)). Similarly, HDAC activity was negatively associated with ATP level (p = 0.0038, R2 = 0.26, Figure 6(e)). However, a significant positive association was found between HDAC activity and H2O2 level (p = 0.0340, R2 = 0.15, Figure 6(f)). Together, these measures suggest that inhibition of HDAC activity is associated with reduced OS and greater ATP production.
We then determined relationships between AH3 level and OS measures. A significant positive correlation existed between AH3 expression level and TAC (p = 0.0185, R2 = 0.18, Figure 6(g)) and between AH3 expression and ATP level (p = 0.0421, R2 = 0.14, Figure 6(h)). However, there was no significant relationship between AH3 expression and 4-HNE adducts (p = 0.1504, R2 = 0.07, data not shown) or H2O2 level (p = 0.6566, R2 = 0.01, data not shown). The trends of interactions with AH3 were opposite in direction from the trends of interaction with HDAC activity, a pattern that is consistent with the negative relationship between the two. Finally, H2O2 correlated negatively with citrate synthase activity (p = 0.0002, R2 = 0.39, Figure 6(i)), supporting an increase in mitochondrial health associated with reduced OS. The strength and number of significant interactions between our measures of OS and HDAC activity or AH3 support a model wherein VOR action and HDAC enzyme activity play a role in OS reduction via both histone- and non-histone mechanisms. If AH3 functioned as the sole mechanistic intermediate between VOR/HDAC activity and downstream effects on OS, we would expect measures of OS to exhibit stronger relationships to AH3 than to HDAC activity.
Sex-specific differences in VOR treatment underlying significant correlations
Consistent with previous observations on HDAC activity (Figure 2(g)) and ATP level (Figure 5(e)), we observed in our regression plots that a number of HDAC- and OS-related measures appeared to cluster by sex (Figure 6). To gain a deeper understanding into how sex and treatment contribute, for a subset of the most highly significant correlations (Figure 6(b)–(i)), we investigated correlative substructures within sex and across treatment (Figure 7). In males, the relationship between ATP and 4-HNE adduct (Figure 7(a)) revealed a wide range of 4-HNE adduct values for the control (grey) and 0.36 mg/g VOR groups (red). Consistent with increased tolerability in 0.18 mg/g group, the 0.18 mg/g group (blue) occupied a lower, more compressed range of 4-HNE adduct values associated with higher ATP levels (Figure 7(a)). A similar pattern was observed in the relationship between AH3 and HDAC activity values (Figure 7(b)), in which the control group (grey) had a larger range than the 0.18 mg/g VOR (blue) or 0.36 mg/g VOR groups, all of which correlated well with AH3 expression values. Finally, distinct spatial relationships were discovered between HDAC activity and ATP levels for control, 0.18 mg/g, and 0.36 groups (Figure 7(c)). In contrast, the same within-drug correlations were less distinct, and there was more spatial overlap between VOR drug groups in females (Figure 7(d)–(f)). Overall, there was a significant sex difference in the robustness of the correlations, as demonstrated by a higher goodness of fit (R2) value in males (Figure 7(g)).
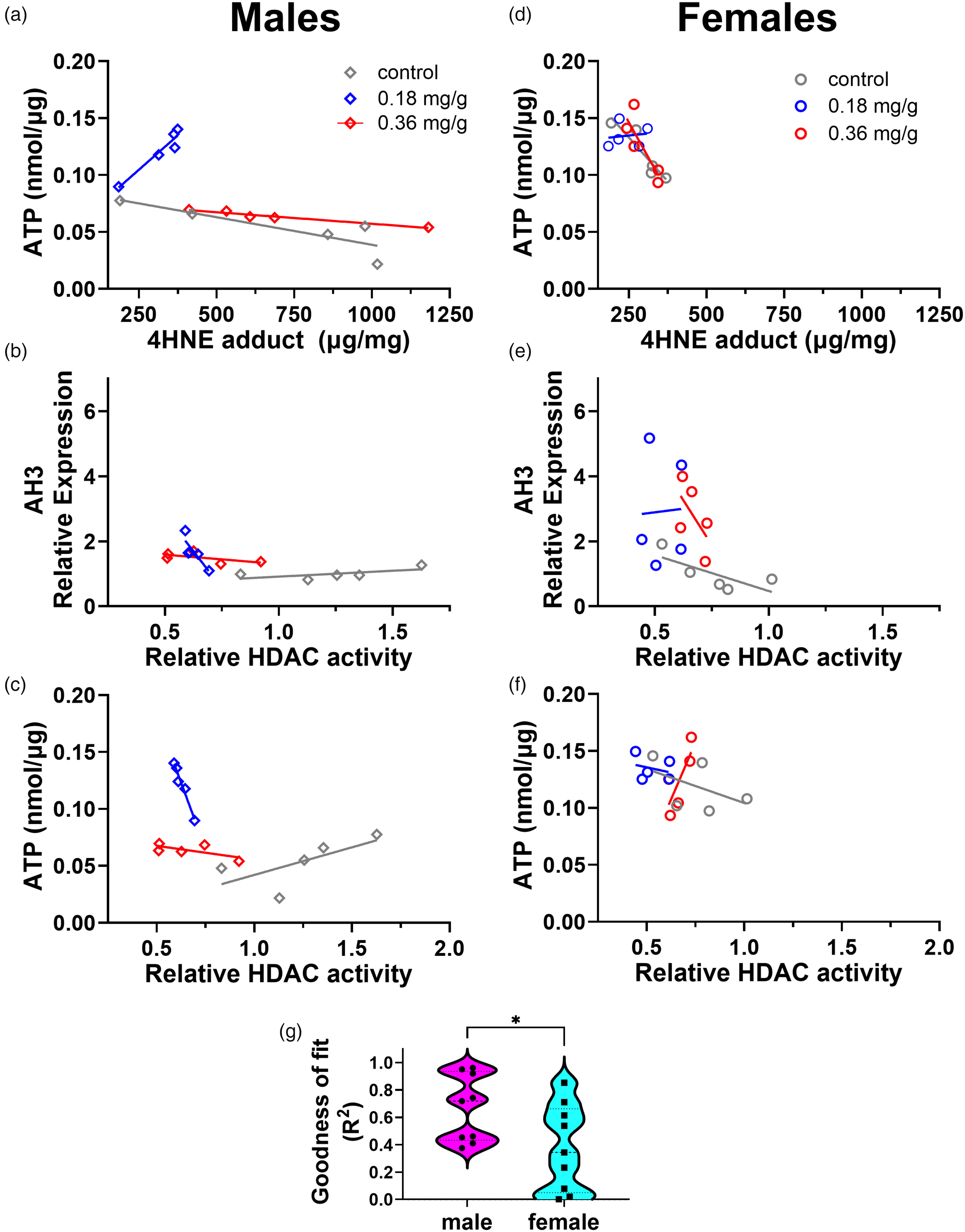
Sex-specific differences in VOR treatment underlying significant correlations. For males, (a) ATP versus 4-HNE adduct, (b) AH3 expression versus HDAC activity, and (c) ATP versus HDAC activity are shown for the three VOR groups. For females (d–f), the same correlations are shown. (g) Graph illustrating significant differences in goodness of fit (R2) of linear regression lines for males and females.
Lower-dose, dietary VOR maintains body weight over an extended time course
Having achieved a tolerable, effective concentration that can cause increased brain histone acetylation in vivo after 14 days of treatment, we then sought evidence that VOR is tolerable over a longer timeframe. Brain HDAC activity was inhibited in the 0.18 mg/g VOR group (Figure 2(g)), but the slightly diminishing diet consumption from week 1 to week 2 presented concerns about tolerability relative to controls (i.e., Figure 1(e)–(g)). To more thoroughly evaluate tolerability, we tested 0.18 mg/g VOR diet for 9 weeks using hAβ-KI mice that were more advanced in age (14–17 months). Body weights were tracked for individual mice in the control and 0.18 mg/g VOR groups (Figure 8(a) and (b)). Overall, compared to initial body weight, no significant changes were observed in body weights at later time points for the control diet (F(2,10) = 0.4573, p = 0.6497). Similarly, body weight was maintained in the 0.18 mg/g VOR group (F(1,9) = 0.8292, p = 0.4562). Percent change in body weight, relative to initial body weight, was also determined for each mouse at each time point (Figure 8(c)). Diet did not significantly affect body weight, and the two diet groups did not differ significantly at any time point when comparing percent change in body weight (F(1,10) = 0.2558, p = 0.6240, p > 0.05 for all timepoints, Figure 8(c)).
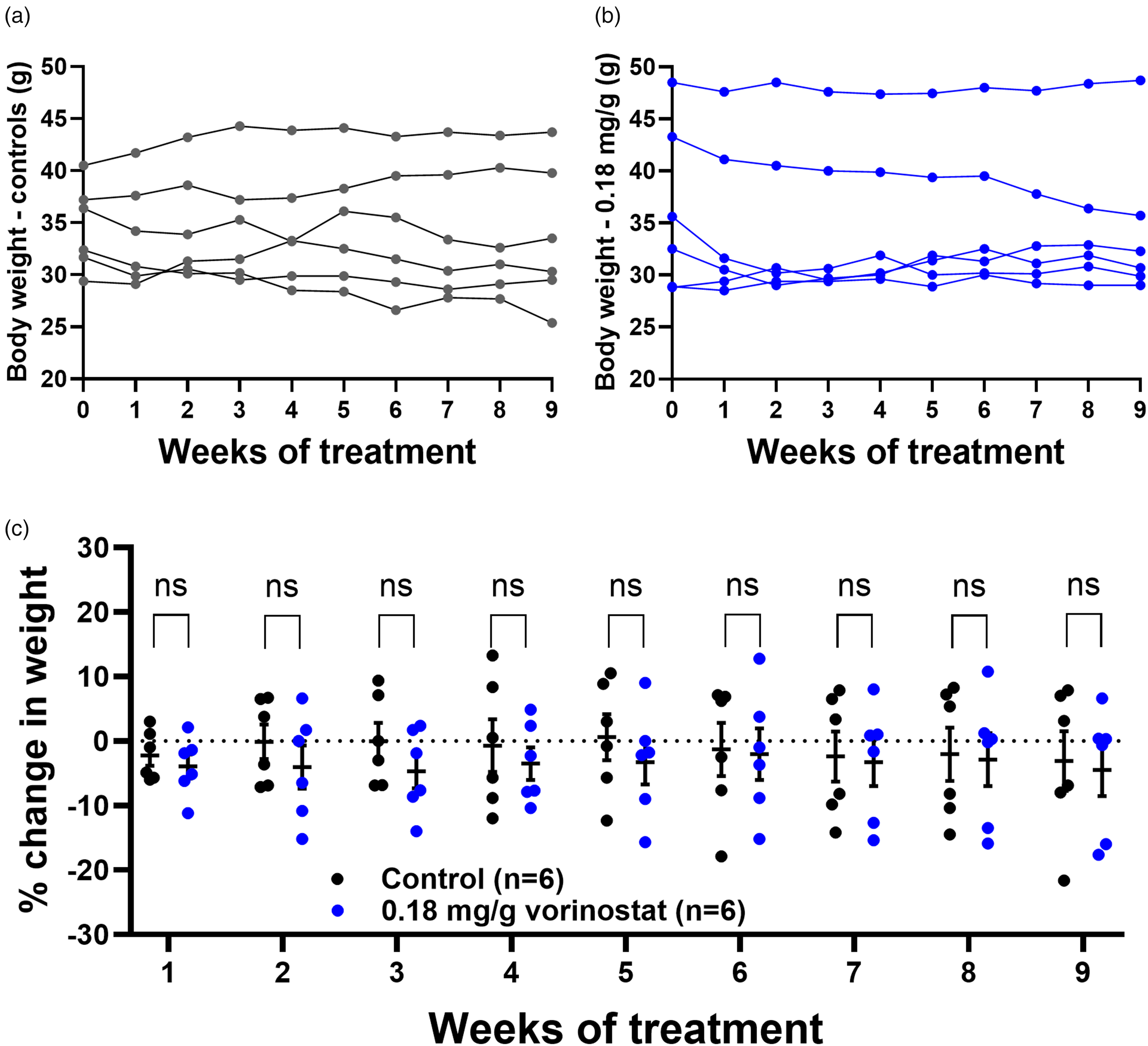
VOR (0.18 mg/g) administered through diet is tolerable over a longer time scale. (a) There was no significant difference between the pretreatment body weights and weights at any later timepoint for either (a) control or (b) 0.18 mg/g VOR diet. (c) Percent change in weight relative to pretreatment weight did not significantly differ between control and 0.18 mg/g VOR diets for up to 9 weeks. ns: not significant.
In summary, our results demonstrate evidence that 0.18 mg/g VOR in diet inhibits HDAC activity and increases brain acetyl-histone H3. The 0.18 mg/g VOR diet was also associated with lower OS measures and higher indicators of mitochondrial health. Finally, we demonstrate that VOR, delivered noninvasively through diet, can be sustained for long-term studies in an AD mouse model.
Discussion
In this study, we administered VOR to 10–12-month-old hAβ-KI mice for 14 days via pelleted diets containing 0.0 mg/g (control), 0.18 mg/g, or 0.36 mg/g VOR. Males in the 0.36 mg/g VOR group failed to demonstrate the same weight gain as the control and 0.18 mg/g groups (Figure 1(d)), implying that the high dose is less tolerable. Both VOR diets increased brain AH3 expression, with larger fold-changes occurring in females (Figure 2(e)). Females were also found to have lower basal HDAC enzymatic activity (Figure 2(g)). Aβ40 and Aβ42 (Figure 3), as well as synaptic proteins (Figure 4) were not significantly affected by diet, but significantly higher levels of Aβ42 were observed in females compared to males in all groups (Figure 3(b)).
We also examined the effects of dietary VOR treatment on markers of OS and mitochondrial health. VOR decreased H2O2 and lipid peroxidation, while antioxidant capacity and markers of mitochondrial heath were increased (Figure 5). The 0.18 mg/g VOR diet was more tolerable and appeared to be more neuroprotective than the 0.36 mg/g diet. An extended test (∼2 months) of 0.18 mg/g VOR diet caused no significant weight loss or other overt toxicities in mice 14–17 months of age (Figure 8), further supporting its use of low-dose VOR in long-term AD mouse studies.
Dietary VOR as a novel route of administration for noninvasively increasing brain histone acetylation levels
Brain AH3 level was significantly increased by VOR diets (Figure 2(e)), demonstrating that VOR can be successfully administered noninvasively via diet, crossing the BBB to target brain HDACs. VOR can also be noninvasively administered via drinking water but is not very soluble in water. The excipient 2-hydroxypropyl-beta-cyclodextrin (HP-β-CD) is used to achieve solubility.27,58,59 Effects of HP-β-CD are not well understood in mice, but several studies in rats have found mild toxicities and histological changes in several organ systems, including liver, bladder, and kidneys, at similar or lower doses than those used to administer VOR in drinking water.60,61 Studies have also paradoxically reported that the cholesterol-binding capability of HP-β-CD confers neuroprotection in models of AD, Parkinson's disease, and Niemann-Pick disease. 62 Thus, the biological effects of HP-β-CD, as well as effects on drug solubility, 63 may complicate studies of AD, particularly in long-term studies.
In contrast to studies that show stable expression of total histone H3 similar to that of housekeeping genes, we found a non-significant trend toward increased H3 expression in VOR-treated animals, whereas β-actin expression was relatively stable across groups. A previous study reported that, relative to non-AD individuals, AD patients have higher levels of both AH3 and total H3 in the temporal cortex, which correlate strongly with each other, 64 suggesting that increased total histone H3 is a component of AD pathology. Our results on individual AH3 and/or H3 expression, when normalized to β-actin expression, are internally consistent with these human findings.
Given the large multi-fold inductions in AH3 observed in some in vitro assays,51,52,65,66 combined with the observed reduction in brain HDAC enzymatic activity with VOR diet (Figure 2(g)), and the observation that AH3 expression correlated negatively with HDAC activity (Figure 6(c)), it is somewhat surprising that we did not observe larger increases in histone acetylation in our VOR-treated mice. This was not likely due to insufficient absorption as vorinostat oral bioavailability is high and not greatly affected by food,67,68 and the calculated delivery was very close to the intended target doses. However, higher basal HDAC enzymatic activity was present in males than in females (Figure 2(g), controls; Figure 6(c)), which may account in part for the smaller increases in AH3 expression achieved in males in the treatment groups (Figure 2(e)).
Antioxidant activity of low-dose VOR
OS has been recognized as an upstream mechanism mediating Aβ overproduction and AD progression.69,70 That low-dose VOR was protective against H2O2-mediated OS (Figure 5(a)) suggests that the underlying mechanisms should be explored for intervention early in AD. Later in AD, neuronal OS may also trigger p53-induced apoptosis, contributing to neuropathogenesis in AD and other neurodegenerative disorders. 71 Prior studies support these findings. In rat PC12 cells, VOR alone increased the antioxidant defense enzyme superoxide dismutase and was synergistically protective against Aβ25–35-induced cytotoxicity when combined with curcumin. 72 In a model of corticosterone-induced stress, VOR increased NRF2 expression and hippocampal GSH levels. 73 Increased NRF2 signaling implies the increased presence of reactive oxygen species (ROS), but may also indicate that, through reacetylation, VOR epigenetically increases transcription of endogenous antioxidant defense-related genes. Interestingly, the dose of 0.18 mg/g appeared to shift the nature of some biological interactions, showing notably different regression slopes from mice on control or 0.36 mg/g diet (Figure 7(a)). For example, on the control and 0.36 mg/g VOR diet, ATP and 4-HNE were negatively correlated; however, in the 0.18 mg/g VOR cohort, ATP and 4-HNE were positively correlated (Figure 7(a)). Future studies are needed to clarify the underlying molecular mechanisms. Overall, our findings that VOR possesses antioxidant-like activity at a low dose is appealing in the context of simultaneously counteracting both underlying HDAC- and OS-mediated AD pathology. These promising results provide a foundation for larger studies that employ a complete, full factorial experimental design.
Vorinostat did not significantly affect soluble Aβ and synaptic markers in hAβ-KI mice
OS is heavily implicated in early AD pathophysiology and has been reported to precede or cause amyloid pathology in mouse and C. elegans models of AD as well as in Down's Syndrome patients, studies which others have reviewed.74,75 There was a trend toward reduction of both Aβ40 and Aβ42 peptide species in the 0.18 mg/g VOR group, but it was not statistically significant. Moreover, the synaptic proteins PSD-95 and synaptophysin were not significantly altered by VOR diet. Baglietto-Vargas and colleagues reported detectable soluble Aβ40 and Aβ42 as early as two months, 42 whereas our mice were 10–12 months of age. Therefore, preventing or reversing amyloidosis may require an earlier and/or extended treatment period. The study of amyloidosis mechanisms in hAβ-KI mice is somewhat limiting because these AD mice do not form Aβ plaques. 42 Nevertheless, if OS and mitochondrial dysfunction are indeed upstream of amyloidogenesis, then low-dose VOR may be more effective in prevention than reversal approaches to treating AD.
Sex differences in response to vorinostat administration
In our study, VOR effects tended to be more prominent in males than females (Figures 6 and 7), which may be partly attributable to increased baseline HDAC activity in males (Figure 2(g)). These sex-specific differences are consistent with several previous studies. For example, following chronic low-dose administration of the HDACi chidamide to 3xTg-AD mice, glucose tolerance and transcriptomic profile changed more significantly in males. 29 Consistent with the blunted effect in females, it was reported that 25 or 50 mg/kg VOR for 14 days did not significantly improve deficits in fear conditioning in a study restricted to female Tg2576 mice. 76
Our study is consistent with a reduced effect of VOR in female hAβ-KI mice. It is possible that differing BBB penetration contributes to sex-specific differences, which could be linked to differing VOR pharmacokinetics and/or differing AD-related pathology associated with BBB degeneration and/or permeability. BBB leakage occurs in many mouse models of AD, 77 although it is unclear to what extent this occurs in hAβ-KI mice. Sex differences in BBB permeability have been reported in mice78,79 as well as humans. 80 Estrogen may help protect BBB integrity from inflammation in mice, 81 but unlike humans, estrogen does not decrease in mice following menopause. 82 However, the literature is inconsistent regarding sex-specific differences in BBB permeability.80,83–85 Therefore, additional research is needed to fully determine the extent to which sex-specific differences in BBB and AD pathology account for sex-specific differences in central VOR actions.
Finally, sex differences in epigenetics have been reported. While genome-wide acetylation of histone H3 lysine 27 (H3K27) did not differ between male and female PS1/APP mice at 13 months of age, methylation of H3K27 residues was increased in females relative to males. 86 Therefore, sex differences in VOR response may not be attributable to global baseline differences in H3 acetylation, but rather to sex differences in methylation. It is possible that the capability of VOR to elicit acetylation changes is affected by histone methylation. Methylation and acetylation are typically mutually exclusive states, and HDACs have been found in complexes with histone methyltransferases, suggesting cooperativity in epigenetic repression. 87
Potential underlying molecular mechanisms of vorinostat action in AD
The observed VOR-induced changes in OS markers may be driven by 1) altered transcriptional regulation caused by epigenetic effects of VOR, 2) direct non-epigenetic antioxidant effects, or 3) a combination of these two causes. Evidence supports direct interactions of OS, HDAC dysregulation, and Aβ. Following exposure of primary mouse neurons to hypoxic conditions in vitro, reduced histone H3 acetylation led to reduced expression of neprilysin, 17 a key enzyme mediating Aβ clearance. 88 Hypoxic conditions can also lead to OS, both directly and indirectly, via mitochondrial dysfunction, Aβ accumulation, and inflammation. 89 Finally, stereotactic injection of Aβ1–40 fibrils in rats increased HDAC2 expression, which decreased AH3 at the neuroligin promoter and neuroligin expression, impairing synaptic transmission and spatial learning. 18 Together, these studies suggest a vicious cycle of Aβ toxicity and epigenetic dysregulation in AD, ultimately leading to homeostatic collapse. Despite beneficial effects on mitochondrial health and OS measures, our results suggest that VOR may not reduce Aβ levels. This outcome is consistent with Benito and colleagues (2015), in which the effects of oral vorinostat were studied in APP/PS1 mice, a model of amyloid deposition. 27 They found that vorinostat increased H4 acetylation, improved memory, restored transcriptomic and splicing changes, and had anti-inflammatory effects. However, these effects were not accompanied by a reduced Aβ plaque load. 27 Corroborating this study is the finding by Cuadrado-Tejedor and colleagues that administration of 12.5 mg/kg VOR IP to Tg2576 mice, an APP model, for 4 weeks resulted in improved learning, reduced cortical pTau, and increased dendritic spine density in CA1 pyramidal neurons relative to vehicle controls, supporting low dose VOR in reducing AD pathology. 37 However, VOR did not reduce full-length APP or Aβ42 levels. 37 Thus, if OS contributes to Aβ pathology, VOR-induced reduction in OS may require earlier and/or longer VOR treatments to reduce Aβ pathology.
VOR may have activities independent of epigenetic effects in the CNS, particularly when considering low doses of VOR and its low level of BBB-barrier penetrance. It is known that Class I/II HDACs can affect lysine acetylation of non-histone proteins, many of which are cytosolic,90–92 and in vitro VOR has been found to increase acetylation of non-histone proteins. 65 One study using genetic cancer mouse models found that at low VOR doses, the anti-inflammatory effects of VOR were more prominent than increases in H3 acetylation, 51 a finding that corroborates the anti-inflammatory effects found by Benito and colleagues. 27 Future work may provide additional context and clarify the extent to which non-epigenetic contributions play roles in potential therapeutic mechanisms.
Conclusions
In conclusion, we have tested two VOR-supplemented diets in an AD mouse model. At the lower dose, reductions of HDAC enzymatic activity, increased AH3 acetylation, reduced OS, and improved mitochondrial health were observed. Although this study did not include behavioral testing, we examined synaptic proteins as a proxy for cognition. Although VOR did not overtly affect either of the synaptic proteins tested, these studies were performed relatively early for the hAβKI mouse model. Future studies are planned for earlier intervention and an extended time course of treatment. However, when considered in the context of prior characterization42,43 and prior studies of vorinostat in amyloid deposition models,27,37 our findings point toward the suitability of 0.18 mg/g VOR diet for long-term prevention studies. While it is unlikely that the short-term interventions performed were of a duration needed to alter the trajectory of disease progression, we found promising molecular and biochemical changes toward neuroprotection, in particular, reduced OS and improved mitochondrial health. It is likely that these changes, achieved across the lifespan, would cumulatively and substantially reduce or prevent neurodegeneration.
Ultimately, AD is best combatted by prevention. We view our results in addressing tolerability and molecular mechanisms as essential for future prophylactic treatment strategies. Oral delivery in preclinical studies should offer better clinical translational potential than other, more invasive routes of administration. Preclinical studies employing diet-based drug administration will help to reduce anxiety, pain, and distress for the animals, potentially allowing for more meaningful behavioral outcomes as well as improving the logistics and cost feasibility of large-scale, long-term prevention studies. Applying this strategy to enable long-term preventive dosing of VOR or other related drugs may facilitate preclinical experiments that better predict clinical outcomes. Although physicians are currently unable to predict future late-onset AD in asymptomatic individuals, VOR may offer improved outcomes to patients with early-stage AD, diagnosed as mild cognitive impairment.
Footnotes
Acknowledgments
We thank Mr. Warren Smith (Dyets, Inc.) for his valuable input on the selection and formulation of the diets. Special thanks to Dr. Kevin Pruitt (University of North Carolina) for his valuable suggestions and early discussions about HDACs and HDAC assays.
ORCID iDs
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by National Institutes of Health grant NIH R01 AG073826 and NIH R01 AG071859 (to JJL) and NIH R01 AA027096 (to IP).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The original data and statistical analysis in this study are available by the authors upon reasonable request.