
Abstract
Background
An 8-week, phase 3b, randomized, placebo-controlled trial demonstrated that pimavanserin, a selective 5HT2A inverse agonist, is generally well tolerated in elderly patients with neuropsychiatric symptoms related to neurodegenerative disease (NDD).
Objective
This open-label extension (OLE) study assessed the long-term safety and tolerability of pimavanserin.
Methods
Patients from the antecedent double-blind (DB) trial who were treated with oral pimavanserin (34 mg/day) or placebo were enrolled. The safety analysis population included all patients who received ≥1 dose of pimavanserin. The primary endpoint was treatment-emergent adverse events (TEAEs). Exploratory endpoints included change from baseline in Extrapyramidal Symptom Rating Scale-Abbreviated (ESRS-A), Mini-Mental State Examination (MMSE), Clinical Global Impression-Severity (CGI-S), EuroQoL 5-Dimension 5-Level (EQ-5D-5L), and Sleep Disorders Inventory (SDI) scores.
Results
Patients (N = 595; mean age, 72.2 years) received pimavanserin treatment (mean exposure, 312.4 days). Most patients (95.3%) had dementia (68.7% of whom had Alzheimer's disease), and 70.6% were concomitantly treated with anti-dementia drugs. TEAEs occurred in 238 (40.0%) patients, and 37 (6.2%) had a serious TEAE; 1 (0.2%) was pimavanserin-related. TEAEs resulted in treatment discontinuation in 39 (6.6%) patients. Fatal TEAEs occurred in 11 (1.8%) patients (none considered related to pimavanserin). The mean (standard error) change from DB baseline to OLE Week 52 in MMSE, ESRS-A, CGI-S, EQ-5D-5L, and SDI scores was +0.9 (0.21), −0.3 (0.22), −1.0 (0.05), + 10.7 (0.87), and −0.9 (0.07), respectively. No patients reported suicidal behavior.
Conclusions
Pimavanserin was generally well tolerated in frail older adults and elderly patients with neuropsychiatric symptoms related to NDD for up to 52 weeks of treatment.
Introduction
Pimavanserin is the only medication approved by the US Food and Drug Administration (FDA) for the treatment of hallucinations and delusions associated with Parkinson's disease psychosis. 1 This study was conducted to further elucidate the long-term safety and tolerability of pimavanserin in a patient population that is highly sensitive to the limiting adverse events (AEs) that are commonly reported for antipsychotic treatment, such as falls, tremor, orthostatic hypotension, somnolence, extrapyramidal symptoms, and death.2–4 In addition, elderly patients often have high rates of polypharmacy, 5 which can increase the number of AEs experienced by patients.
Neuropsychiatric symptoms in patients with neurodegenerative disease (NDD) frequently include anxiety, agitation, apathy, motor disturbances, psychosis, hallucinations, delusions, and disinhibition. 6 Symptoms can appear at any stage of the NDD and will often fluctuate in frequency and severity.6,7 Approximately 30% to 60% of patients with Parkinson's disease experience neuropsychiatric symptoms, and almost all patients experience at least 1 neuropsychiatric symptom throughout the disease course.8,9 Psychosis is a common neuropsychiatric symptom in patients with NDD, particularly in neurodegenerative dementia, with recent estimates suggesting it affects 20% to >50% of patients depending on the type of NDD. 10
Given the progressive nature of NDDs, the associated neuropsychiatric symptoms often negatively impact health-related quality of life and activities of daily living and increase caregiver burden.6–8
Treatment with pimavanserin is not associated with motor dysfunction or cognitive decline. Pimavanserin is a selective 5-HT2A receptor inverse agonist and, to a lesser extent, a 5-HT2C receptor inverse agonist/antagonist. 11 Pimavanserin has negligible affinity for dopaminergic, muscarinic, histaminergic, and adrenergic receptors. 11 Due to this high degree of specificity, pimavanserin lacks many of the AEs associated with those receptors. 2
In a preceding phase 3b, randomized, double-blind (DB), placebo-controlled, 8-week study (NCT03575052), which enrolled frail older adults and elderly patients with neuropsychiatric symptoms related to NDD, pimavanserin was generally safe and well tolerated. 12 Additionally, pimavanserin was not associated with motor symptoms or cognitive impairment.
Given that patients with neuropsychiatric symptoms related to NDD, including Parkinson's disease, are highly sensitive to AEs associated with antipsychotics, it is important to understand the long-term safety profile of pimavanserin in patients with NDD beyond 8 weeks of treatment to inform the treatment of patients with Parkinson's disease psychosis. The objective of this open-label extension (OLE) study was to expand on the results from the antecedent study to evaluate the safety and tolerability of long-term pimavanserin treatment in frail older adults and elderly patients with neuropsychiatric symptoms related to NDD.
Methods
Study design and participants
In this 52-week, multicenter, phase 3b, OLE study (NCT03623321), adult and elderly patients with neuropsychiatric symptoms related to NDD received pimavanserin 34 mg once daily. Patients who completed the antecedent DB study of pimavanserin were eligible to enroll. A full description of the antecedent trial design and results have been previously reported in detail. 12 Key inclusion criteria of the antecedent trial included patients being ≥60 years old, requiring some or complete assistance with instrumental or basic activities of daily living, meeting all clinical criteria for a NDD, and having neuropsychiatric symptoms severe enough to warrant antipsychotic treatment. All patients who received either pimavanserin or placebo treatment during the antecedent study were eligible to enroll in this OLE extension. The study timeline consisted of an open-label treatment period for 52 weeks and a safety follow-up period with a telephone call to the patient and caretaker conducted 30 (+4) days after the last dose of pimavanserin. Eligible patients included those who satisfied all entry criteria for the antecedent pimavanserin study if the investigator determined they may benefit from longer-term therapy with pimavanserin. Patients who completed the antecedent study were required to have a designated care partner to support study completion and adherence with study procedures. Female patients could not be pregnant or breastfeeding. The main exclusion criteria were patients receiving hospice or end-of-life palliative care or having a heart rate <50 beats per minute, abnormal electrocardiogram (ECG), having suicidality at baseline, and having any disease or disorder that would increase the risks associated with pimavanserin or interfere with the study. In addition, concomitant treatment with medications that prolong the QT interval were prohibited or restricted during the study.
This study was conducted according to the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice Guidelines, and local regulatory requirements. The protocol was reviewed and approved by an Institutional Review Board or Ethics Committee at each site. All patients provided informed consent prior to study participation.
Procedures
Eligible patients received pimavanserin 34 mg once daily for 52 weeks. Dose adjustments to pimavanserin 20 mg or back to pimavanserin 34 mg were allowed at any study visit after the baseline visit and were based on the investigator's judgment. Clinic visits were conducted at baseline and Weeks 2, 4, 8, 12, 16, 28, 40, and 52 following the end of treatment from the antecedent pimavanserin study. Pimavanserin was dispensed to the patient at the baseline visit and at each subsequent visit. Assessments performed at the end-of-treatment visit in the antecedent study served as baseline evaluations for this OLE study.
Outcomes
The primary endpoint was safety as measured by treatment-emergent adverse events (TEAEs). An AE was defined as a TEAE if it started on or after the first dose of pimavanserin and ≤30 days after the last dose of pimavanserin in the OLE study. Adverse events reported on Day 1 from predose findings were not considered TEAEs. Exploratory safety endpoints included the Mini-Mental State Examination (MMSE) to measure cognitive function, 13 the Extrapyramidal Symptom Rating Scale-Abbreviated (ESRS-A) to measure changes in extrapyramidal symptoms, 14 and the Columbia-Suicide Severity Rating Scale (C-SSRS) to measure the risk for suicidality. 15 If the patient was not reliable to complete the C-SSRS per the study investigator's judgment, then the Global Clinician Assessment of Suicidality (GCAS) was used. The GCAS is a clinician-rated, 5-point scale of an individual's suicidality. The possible ratings are 0 (“Absent”), 1 (“Feels life is not worth living”), 2 (“Wishes s/he were dead or any thoughts of possible death to self”), 3 (“Suicidal ideas or gesture”), or 4 (“Attempt at suicide”), which are based on the classification of suicidal ideation in older patients with depression, anxiety, and at-risk alcohol use. 16 Patients with a GCAS score of 3 or 4 were considered to be suicidal. 17
The C-SSRS, MMSE, and ESRS-A were assessed at baseline and at each subsequent study visit. Other safety assessments included AEs, vital signs, 12-lead ECG, and standard clinical laboratory tests. Exploratory efficacy endpoints included change from baseline in the Clinical Global Impression-Severity (CGI-S) score for neuropsychiatric symptoms, 18 the EuroQoL 5-Dimension 5-Level (EQ-5D-5L) instrument to assess health-related quality of life, 19 and the Sleep Disorders Inventory (SDI) score to measure sleep disturbances. 20 The CGI-S was measured at baseline and at each subsequent study visit; EQ-5D-5L and SDI scores were measured at baseline and Weeks 12, 28, and 52.
Statistical analysis
Targeted enrollment was up to 750 patients and determined by the number of patients who transitioned from the antecedent study. The enrolled patients included all patients who signed the informed consent, excluding rollover failures
Results
Patient disposition and baseline characteristics
This OLE study was conducted from July 2018 to May 2023. Of the 784 patients from the antecedent study, 596 patients were enrolled, not including 10 rollover failures (Figure 1). After the baseline visit, 1 patient was lost to follow-up and not included in the safety analysis population (N = 595). Of those enrolled, 143 (24.0%) patients terminated the study early and 76% of patients completed the study. The most common reasons for early termination included other (7.2%), adverse event (5.4%), and withdrawal of consent (4.9%).
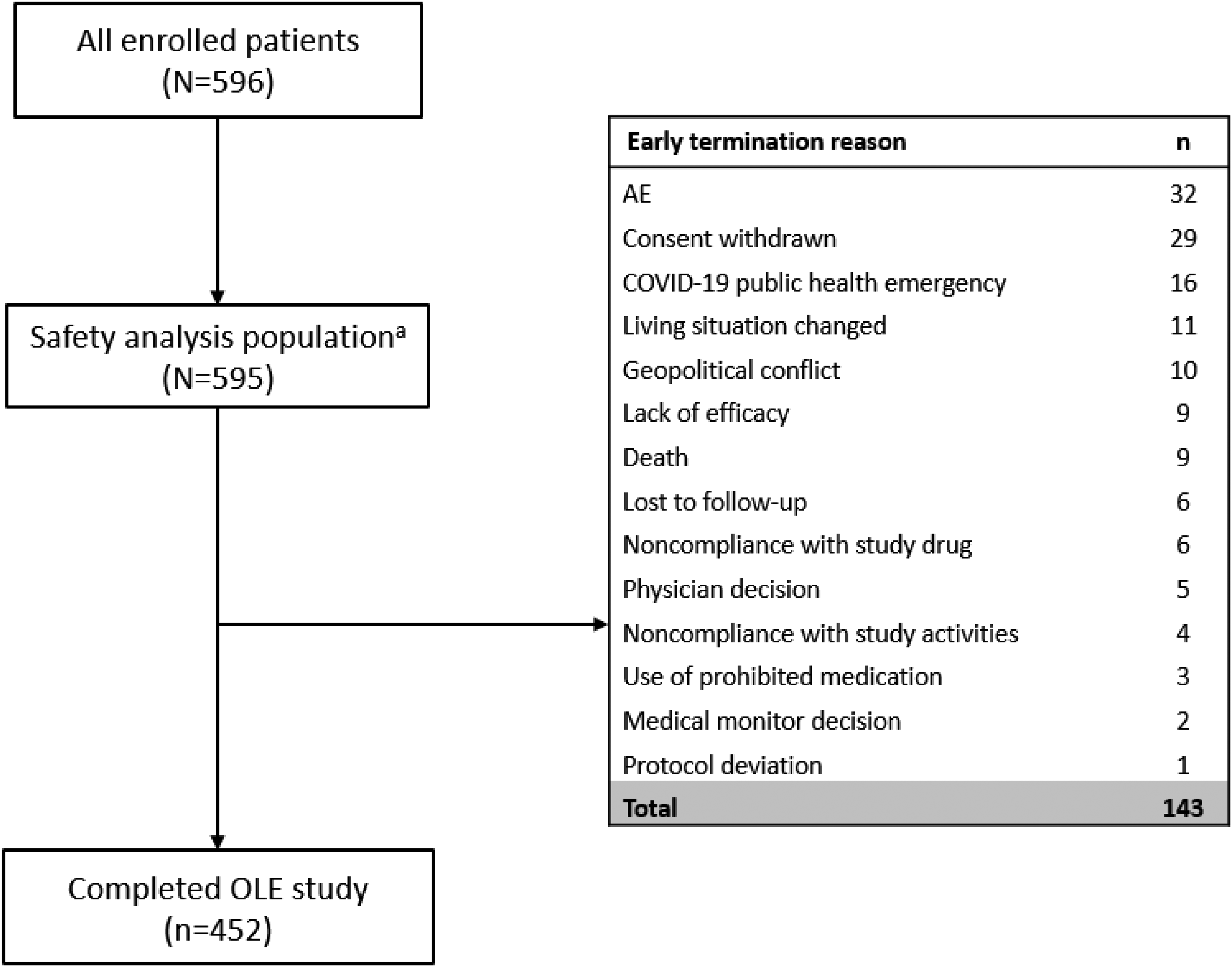
Patient disposition. AE: adverse event; OLE: open-label extension. aPatients who received ≥1 dose of pimavanserin during the OLE period.
Patient baseline demographic and clinical characteristics are presented for the safety analysis population (Table 1). The mean (SE) age was 72.2 (0.27) years, and 58.5% of patients were female. Most patients had dementia and not Parkinson's disease. Alzheimer's disease was the most common dementia subtype (68.7%). At baseline, mean (SE) CGI-S, EQ-5D-5L, and SDI scores were 4.0 (0.03), 61.5 (0.73), and 0.6 (0.04), respectively.
Baseline characteristics (safety analysis population).

Two patients had Parkinson's disease that was not the primary cause of dementia.
Includes patients with Parkinson's disease with and without dementia.
End-of-study visit for antecedent randomized controlled trial.
CGI-S: Clinical Global Impression-Severity; C-SSRS: Columbia-Suicide Severity Rating Scale; ESRS-A: Extrapyramidal Symptom Rating Scale-Abbreviated; EQ-5D-5L: 5-level version of EQ-5D; GCAS: Global Clinician Assessment of Suicidality; MMSE: Mini-Mental State Examination; NDD: neurodegenerative disease; QTcF: corrected QT interval using Fridericia's correction method; SDI: Sleep Disorders Inventory.
In the OLE treatment period, overall mean (SE) duration of exposure to pimavanserin was 312.4 (4.42) days (Table 2). The mean (SE) duration of exposure to pimavanserin by treatment arm in the antecedent double-blind study was similar (placebo, 305.1 [6.60] days; pimavanserin, 319.8 [5.85] days). The mean (SE) average daily dose for all patients was 33.3 (0.1) mg. A total of 34 (5.7%) patients were reduced to 20 mg during the OLE study, of whom 29 were receiving this dose at the last study visit.
Duration of pimavanserin exposure (OL study safety analysis population).a
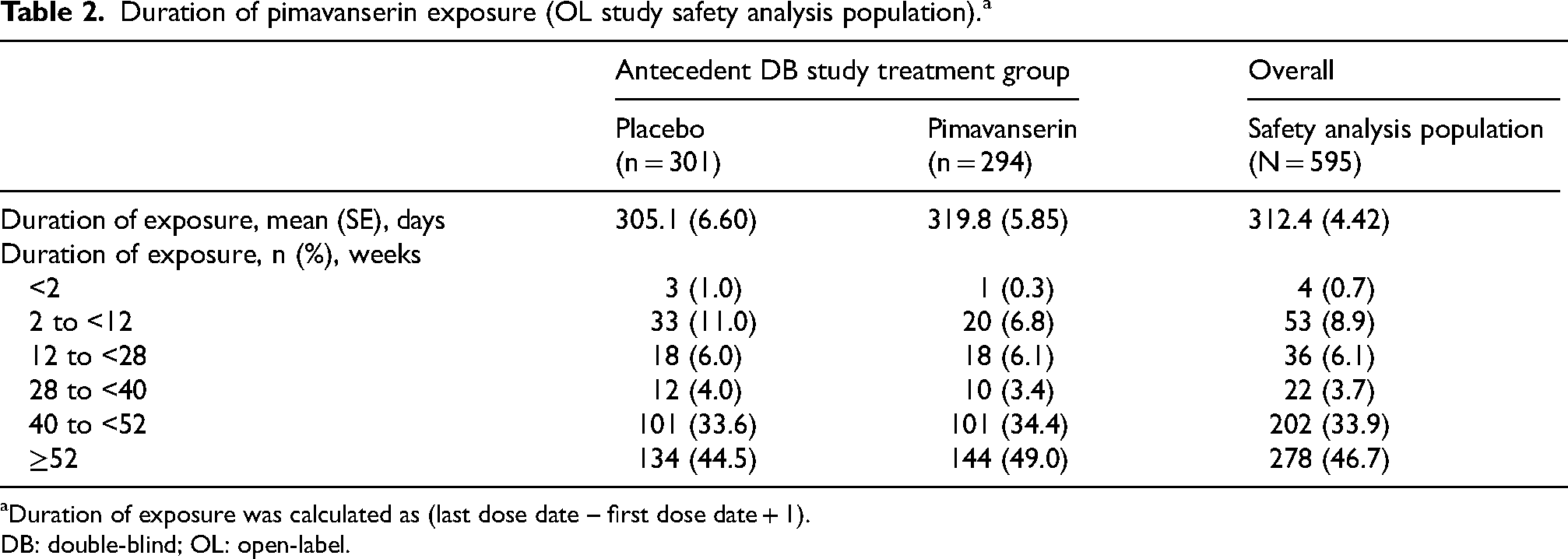
Duration of exposure was calculated as (last dose date – first dose date + 1).
DB: double-blind; OL: open-label.
Nearly all patients (569/595 [95.6%]) were treated with concomitant medications while receiving pimavanserin (Supplemental Table 1). The most frequent concomitant medications were anti-dementia drugs (420/595 [70.6%]), lipid-modifying agents (239/595 [40.2%]), and antithrombotic agents (182/595 [30.6%]).
Safety
Among patients in the OLE safety analysis population, ≥1 TEAE was experienced by 238 (40.0%) patients (Table 3). The frequency of TEAEs in each treatment arm of the antecedent double-blind study was similar (placebo, 110/301 [36.5%]; pimavanserin, 128/294 [43.5%]). Most TEAEs were mild to moderate in severity. The most commonly reported TEAEs included urinary tract infection (4.9%), headache (3.7%), and dizziness (3.4%; Table 4). Among patients who were in the placebo treatment arm of the antecedent double-blind study, the most commonly reported TEAEs were urinary tract infection, headache, and insomnia (3.3% each). Among patients who were in the pimavanserin treatment arm, the most commonly reported TEAEs were urinary tract infection (6.5%), headache (4.1%), and dizziness (3.7%). No TEAEs of orthostatic hypotension or sedation were reported. Falls and tremors were reported in 2.2% and 1.2% of patients in the OLE study, respectively. Additionally, TEAEs of increased weight (0.2%), decreased weight (2.0%), and abnormal loss of weight (0.2%) occurred infrequently. Among the 37 (6.2%) patients with serious TEAEs, the most common serious TEAE was cerebrovascular accident, which was reported in 3 (0.5%) patients (Table 4). Serious TEAEs that only occurred in 1 patient included acute respiratory failure, aggression, anxiety disorder, arteriosclerosis, bradycardia, cardiac arrest, chronic obstructive pulmonary disease, coma, COVID-19 pneumonia, fall, femoral neck fracture, foot fracture, gastrointestinal hemorrhage, head injury, humerus fracture, hypotension, inguinal hernia, ischemic stroke, left ventricular failure, lip and/or oral cavity cancer, myocardial infarction, necrotizing pneumonia, osteoarthritis, Parkinson's disease, perirectal abscess, pulmonary edema, pulmonary embolism, radius fracture, seizure, sepsis, subdural hematoma, syncope, transient ischemic attack, urinary retention, and urinary tract infection. One (0.2%) patient experienced a study drug-related serious TEAE (a fall). A total of 39 (6.6%) patients experienced TEAEs leading to discontinuation, 4 of which occurred in ≥2 patients (prolonged cardiac QT interval, 5 [0.8%]; brain edema, 2 [0.3%]; cerebrovascular accident, 2 [0.3%]; renal impairment, 2 [0.3%]; Table 3). Fatal TEAEs occurred in 11 (1.8%) patients (Table 3), but none were considered to be related to pimavanserin. Therefore, treatment with pimavanserin is not anticipated to increase the risk of mortality.
Summary of TEAEs (OL study safety analysis dataset).
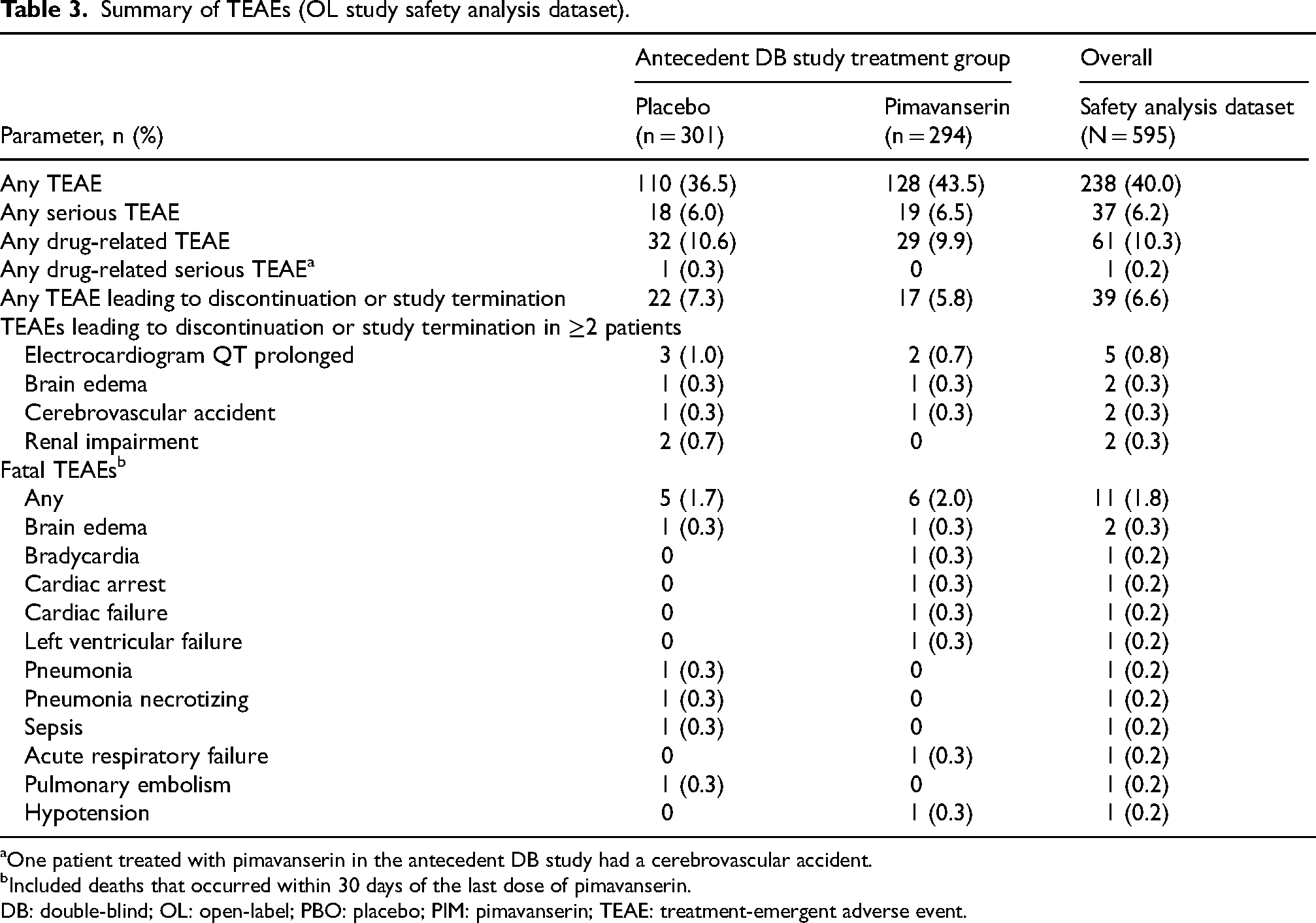
One patient treated with pimavanserin in the antecedent DB study had a cerebrovascular accident.
Included deaths that occurred within 30 days of the last dose of pimavanserin.
DB: double-blind; OL: open-label; PBO: placebo; PIM: pimavanserin; TEAE: treatment-emergent adverse event.
TEAEs reported in ≥2% of patients and any serious TEAEs in >1 patient in the OL study safety analysis population.
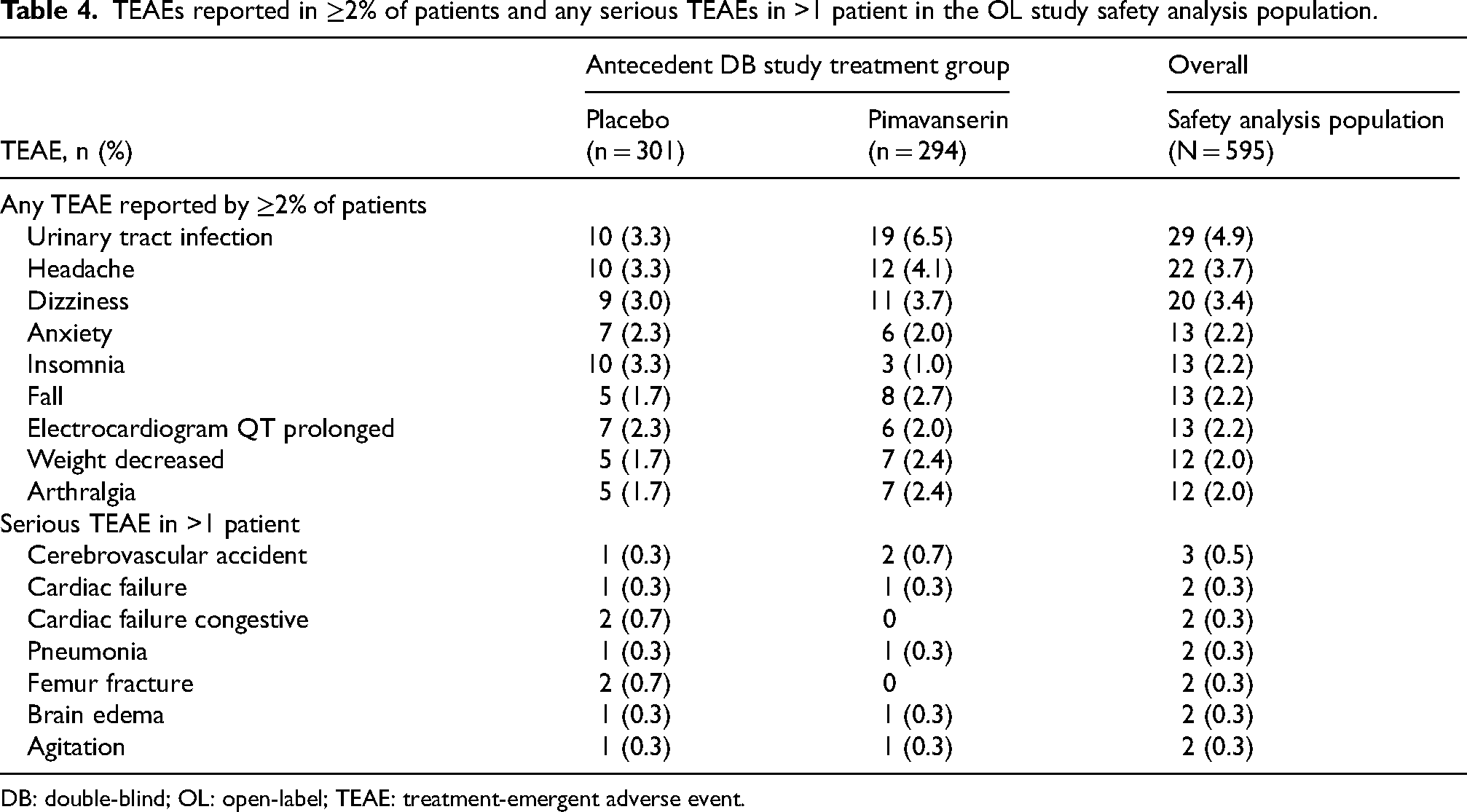
DB: double-blind; OL: open-label; TEAE: treatment-emergent adverse event.
No clinically meaningful changes were observed in the laboratory findings, vital signs, or physical findings. Thirteen (2.2%) patients experienced a TEAE of QT prolongation, 7 [2.3%] of whom received placebo and 6 [2.0%] of whom received pimavanserin in the antecedent double-blind study. In the safety analysis population, >97% of patients had a QT interval ≤450 ms at all study visits, and no patient had an interval >500 ms. The largest mean (SE) increase from OLE baseline in QT interval was 4.8 (1.0) ms at OLE Week 28 in patients who were in the placebo treatment group in the antecedent double-blind study (Supplemental Table 2).
Exploratory safety endpoints
In the safety analysis population, 550/595 (92.4%) and 115/595 (19.3%) patients were assessed with C-SSRS and GCAS, respectively. No reports of attempted suicide or suicidal ideation was reported among patients who were assessed with GCAS. Suicidal ideation was reported in 6 patients as measured by the C-SSRS, and none of these events were considered a TEAE. Of the 6 patients with suicidal ideation, 4 reported the least severe suicidal ideation (wish to be dead), and 2 reported nonspecific active suicidal thoughts. Additionally, no patients reported suicidal behavior as measured by the C-SSRS. Overall, there was little to no change from DB baseline in MMSE score through OLE Week 28 and then it trended downward through OLE Week 52 (Figure 2A). At OLE Week 52, the observed mean (SE) change from the DB baseline was 0.9 (0.21). One patient had a mild cognitive disorder TEAE, but this was not considered to be related to pimavanserin.
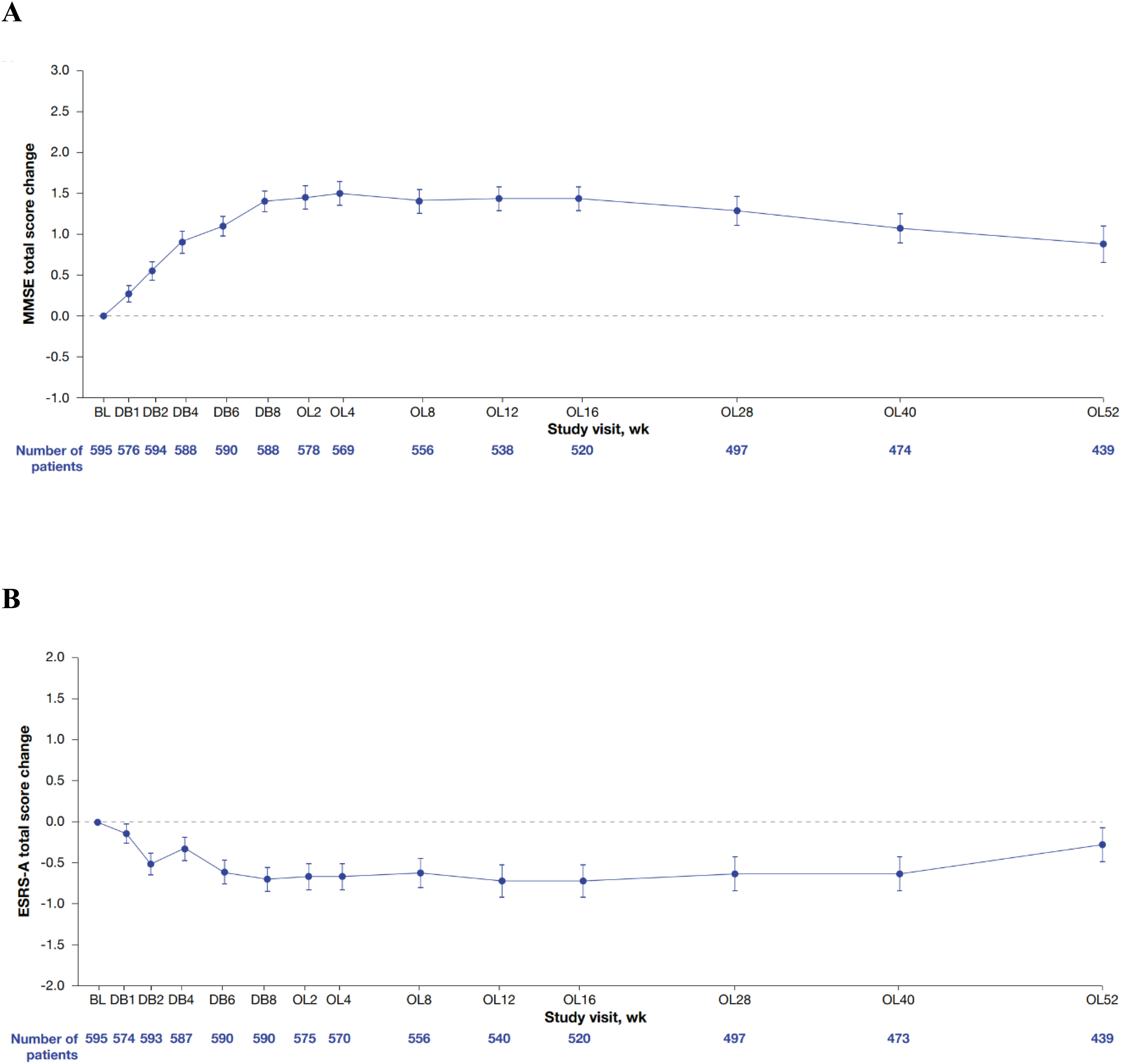
Change from the DB baseline in (A) MMSE score and (B) ESRS-A score. Error bars indicate standard error. BL: baseline; DB: double-blind; ESRA-A: Extrapyramidal Symptom Rating Scale-Abbreviated; OL: open-label; MMSE: Mini-Mental State Examination; PBO: placebo; PIM: pimavanserin.
Similarly, there was minimal change from DB baseline in ESRS-A score for extrapyramidal symptoms through OLE Week 28 and then it trended upward through OLE Week 52 (Figure 2B). At OLE Week 52, the mean (SE) change from the DB baseline was −0.3 (0.22).
Exploratory efficacy endpoints
The CGI-S score for neuropsychiatric symptoms decreased from baseline (Figure 3A). At OLE Week 52, the mean (SE) change from DB baseline was −1.0 (0.05), which corresponded to a 22.2% decrease. In general, the EQ-5D-5L score for health-related quality of life trended upward during the study (Figure 3B). At OLE Week 52, the mean (SE) change from DB baseline was +10.7 (0.87), which corresponded to a 21.6% increase. The SDI score for sleep disturbance decreased from baseline to Week 12 and then remained steady through Week 52 (Figure 3C). At OLE Week 52, the mean (SE) change from DB baseline was −0.9 (0.07), which corresponded to a 66.7% decrease.
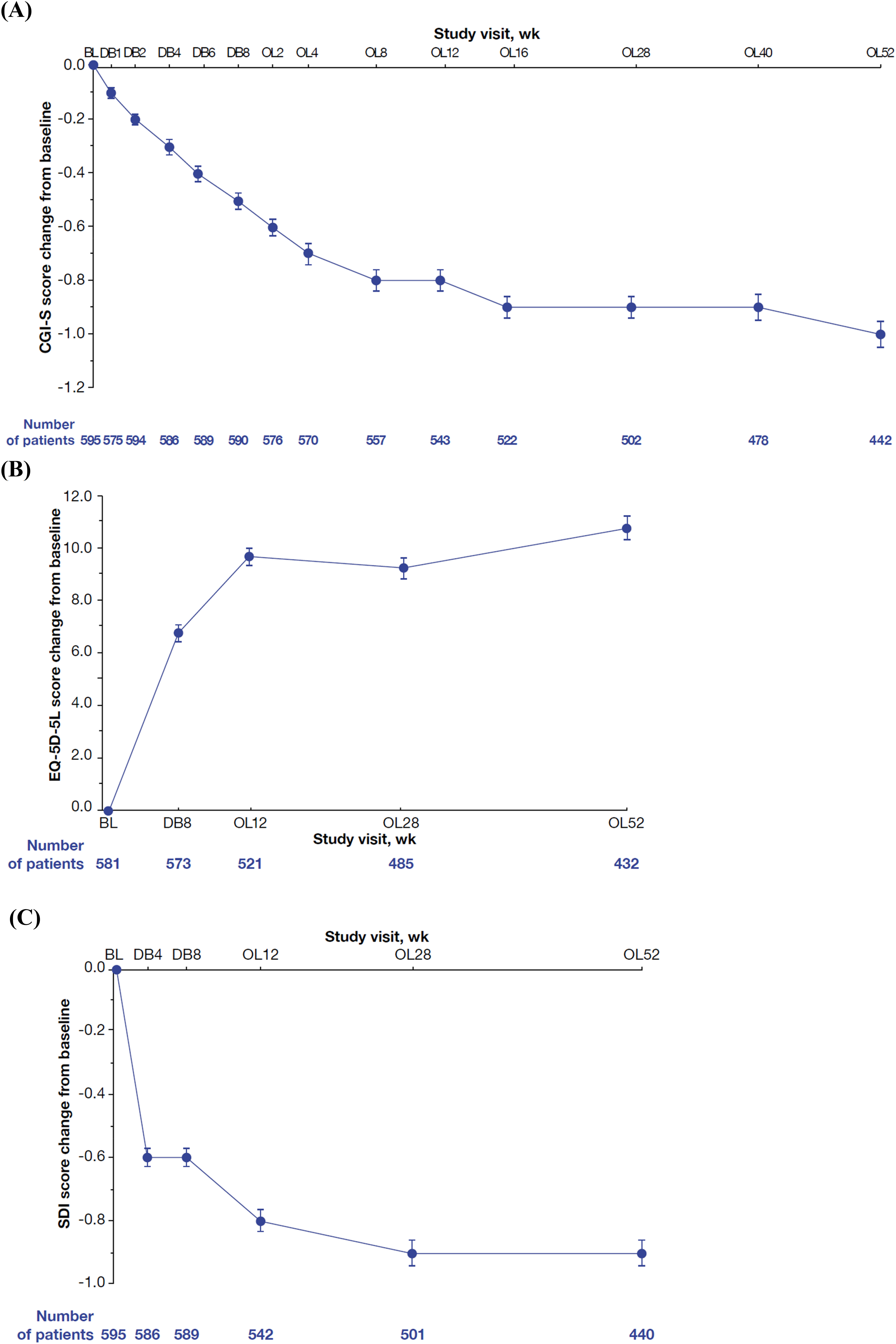
Change from the DB baseline in (A) CGI-S score for neuropsychiatric symptoms, (B) EQ-5D-5L score, and (C) Sleep Disorders Inventory score (safety analysis population). Lower CGI-S and SDI scores indicate less impairment and decreased severity of sleep disturbance, respectively. Higher EQ-5D-5L scores indicate improved health-related quality of life. Error bars indicate standard error. CGI-S: Clinical Global Impression-Severity; EQ-5D-5L: 5-level version of EQ-5D; OLE: open-label extension; SDI: Sleep Disorders Inventory.
Discussion
In this OLE study, treatment with pimavanserin 34 mg once daily in older adults and frail elderly patients with neuropsychiatric symptoms related to NDD was generally well tolerated with no new safety concerns associated with long-term treatment. Among patients in the safety analysis dataset, serious TEAEs and TEAEs leading to treatment discontinuation were only experienced by 6.2% and 6.6% of patients overall, respectively, which highlights the tolerability of pimavanserin. As further evidence of that favorable tolerability, 76.0% of treated patients completed the 52-week study, with 5.7% of patients needing dose reduction. The most frequently reported TEAEs were generally mild to moderate in severity and included urinary tract infection, headache, and dizziness. Of note, TEAEs that are frequently associated with antipsychotics (e.g., orthostatic hypotension, sedation, falls, tremors)2–4 were infrequent or not reported in this 52-week study. The proportion of patients in this study who had falls (2.2%) was much lower than that in a previous 11-year OLE study evaluating the long-term safety and tolerability of pimavanserin with over 400 patients with psychotic symptoms consistent with Parkinson's disease psychosis, which reported that 32.0% of patients had falls. 21 Similarly, in the phase 2 trial of pimavanserin, 21/90 (23.3%) patients treated with pimavanserin experienced a fall adverse event. 22
Findings from this OLE study are consistent with previous studies on the well-characterized safety profile of pimavanserin and help to further inform the long-term safety and use of pimavanserin.12,21–23 The previous phase 3b, 8-week study demonstrated that treatment with pimavanserin was generally safe and well tolerated and not associated with motor or cognitive impairment in frail older adults and elderly patients with neuropsychiatric symptoms related to NDD. 12 Likewise, the phase 2, placebo-controlled, double-blind study of pimavanserin in patients with Alzheimer's disease psychosis demonstrated that pimavanserin has an acceptable tolerability profile and did not have any negative effect on cognition. 22 Results from this 52-week study were similar; pimavanserin was generally well tolerated with no new safety signals.
Given that all antipsychotics, including pimavanserin, contain a boxed warning for elderly patients in the US FDA prescribing label, 1 it is important to understand the mortality risk associated with pimavanserin. In this study, there were 11 deaths, none of which were associated with pimavanserin. In a retrospective analysis of mortality and clinical characteristics among patients who received pimavanserin with clinically diagnosed Parkinson's disease, pimavanserin was not associated with an increased risk of death when compared with patients treated with other off-label atypical antipsychotics, and patients who received pimavanserin had a lower risk of mortality when compared with patients who were untreated.24–26 Although this OLE study did not have a comparator, the data support pimavanserin as a safe and well-tolerated treatment for Parkinson's disease psychosis with or without dementia, with no observed increase in the risk of mortality in the elderly patient population.
Other safety findings from this study were consistent with the well-characterized safety profile of pimavanserin. The mean change from baseline in extrapyramidal symptoms as measured by the ERSA-A was small. Consistent with the phase 2 trial of pimavanserin in patients with Alzheimer's disease psychosis, no cognitive decline or motor dysfunction was associated with pimavanserin. 22 Additionally, the rates of QT prolongation were similar to the rates observed in prior studies of pimavanserin, with patients in this OLE study generally showing a prolonged QT interval after treatment.21,22,24 Moreover, the mean (SE) duration of prolongation in this OLE study (4.8 [1.0] ms) was consistent with the current product label for pimavanserin as well as a previous phase 3 study of pimavanserin in patients with dementia-related psychosis (5.4 [0.9] ms).1,27 No patients reported suicidal behavior. Treatment with pimavanserin was not associated with clinically meaningful changes in laboratory findings, vital signs, or physical findings. Weight gain and metabolic TEAEs occurred infrequently.
Change from baseline in exploratory endpoints measured by CGI-S, SDI, and EQ-5D-5L scores suggests that the beneficial therapeutic effects associated with pimavanserin, observed in the previous placebo-controlled study, 12 may extend beyond Week 8 and continue up to Week 52.
Taken together, these results increase the overall confidence in the long-term safety and tolerability of pimavanserin in older adult and frail elderly patients with neuropsychiatric symptoms related to NDD. It is important to highlight that this population is often more sensitive to the limiting AEs associated with the treatment of atypical antipsychotics, such as falls, somnolence, weight gain, extrapyramidal symptoms, and death.2–4
Although the study included a large sample size from multiple global regions, selection bias may have occurred from the nonrandom selection of patients from the antecedent study. Strengths of this study include the extended trial time of 52 weeks, which provided long-term outcome data on pimavanserin. However, the study was a single-arm, OLE design that lacked a comparator arm, which does not allow definitive efficacy conclusions and limits the safety conclusions that can be drawn.
Conclusion
In this study, pimavanserin was generally well tolerated in older adult and frail elderly patients with neuropsychiatric symptoms related to NDD, including Parkinson's disease (with or without dementia), dementia with Lewy bodies, frontotemporal degeneration spectrum disorders, all-cause dementia with possible or probable Alzheimer's disease, or vascular dementia, for up to 52 weeks of treatment, and no new safety signals were observed. During the study, the exploratory endpoints of change from baseline in neuropsychiatric symptoms, health-related quality of life, and sleep suggest a potential beneficial therapeutic effect of pimavanserin, shown by CGI-S score decrease, EQ-5D-5L score increase, and SDI score decrease. Furthermore, pimavanserin was not associated with cognitive impairment or motor dysfunction. These data further inform and support the long-term use of pimavanserin in patients with Parkinson's disease psychosis with or without dementia.
Supplemental Material
sj-docx-1-alz-10.1177_13872877251345162 - Supplemental material for Pimavanserin safety in adult and elderly patients with neuropsychiatric symptoms related to neurodegenerative disease: An open-label extension study
Supplemental material, sj-docx-1-alz-10.1177_13872877251345162 for Pimavanserin safety in adult and elderly patients with neuropsychiatric symptoms related to neurodegenerative disease: An open-label extension study by Wiesław J Cubała, Ana Berrio, Katherine Chi-Burris, Gustavo Alva, Lambros Chrones and Sanjeev Pathak in Journal of Alzheimer's Disease
Footnotes
Acknowledgements
Medical writing support was provided by Makaila Wallin, PharmD, and Alexander Simon, for Citrus Scientific, a Citrus Health Group, Inc., company (Chicago, Illinois) in accordance with Good Publication Practices guidelines. This support was funded by Acadia Pharmaceuticals.
Ethical considerations
This study was conducted according to the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice Guidelines, and local regulatory requirements. The protocol was reviewed and approved by an Institutional Review Board or Ethics Committee at each site.
Consent to participate
All patients provided written informed consent prior to study participation.
Author contributions
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Acadia Pharmaceuticals Inc.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Data availability statement
Clinical study documents are confidential and the property of Acadia Pharmaceuticals. The study protocol and statistical analysis plan are available upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.