
Abstract
A conundrum in Alzheimer's disease (AD) is why the long-term use of acetylcholinesterase (AChE) inhibitors, intended for treatment of dementia, results in slowing neurodegeneration in the cholinergic basal forebrain, hippocampus, and cortex. The phospho-tau cascade hypothesis presented here attempts to answer that question by unifying three hallmark features of AD into a specific sequence of events. It is proposed that the hyperphosphorylation of tau protein leads to the AD-associated deficit of nerve growth factor (NGF), then to atrophy of the cholinergic basal forebrain and dementia. Because the release of pro-nerve growth factor (pro-NGF) is activity-dependent and is controlled by basal forebrain projections to the hippocampus and cortex, our hypothesis is that AChE inhibitors act by increasing acetylcholine-dependent pro-NGF release and, thus, augmenting the availability of mature NGF and improving basal forebrain survival. If correct, improved central nervous system-selective AChE inhibitor therapy started prophylactically, before AD-associated basal forebrain atrophy and cognitive impairment onset, has the potential to delay not only the onset of dementia but also its rate of advancement. The phospho-tau hypothesis thus suggests that preventing hyperphosphorylation of tau protein, early should be a high priority as a strategy to help reduce dementia and its associated widespread social and economic suffering.
Keywords
Introduction
Decades of research have shown that the amyloid cascade hypothesis of Alzheimer's disease (AD), as originally proposed, 1 is not sufficient to produce successful therapeutic strategies.2,3 Rather, AD is a highly complex multifactorial disease with different subtypes and trajectories.4–7 The newly revised biological definition of AD (NIA-AA criteria) includes abnormal depositions of amyloid-β, hyperphosphorylated tau, and disease-specific patterns of neurodegeneration as its primary biomarkers, 3 in addition to findings of vascular changes and neuroinflammation. 2 Amyloidosis, 8 tauopathy,7,9,10 inflammation, 11 impaired nerve growth factor (NGF) signaling,12–14 and neurodegeneration of the cholinergic basal forebrain appear many years before the onset of dementia. Regardless of the yet-to-be-identified cause(s) of AD, all cases result in dementia, a final late-stage symptom. 15
As shown in Figure 1, the phospho-tau cascade hypothesis combines three biomarkers of AD into a final common pathway to dementia. 16 Specifically, it proposes that hyperphosphorylated tau (phospho-tau)9,17 blocks the retrograde transport of the key neurotrophic factor, NGF.12,13,18 Subsequently, that loss of NGF causes neurodegeneration of the cholinergic basal forebrain 19 and, finally, the cascade ends in dementia. 16 The hyperphosphorylation of tau protein, which can be detected many years before the onset of dementia,7,9 has been proposed as the initiating factor for sporadic disease,17,20 but it is also the proposed triggering event in the phospho-tau cascade to dementia (Figure 1). 16
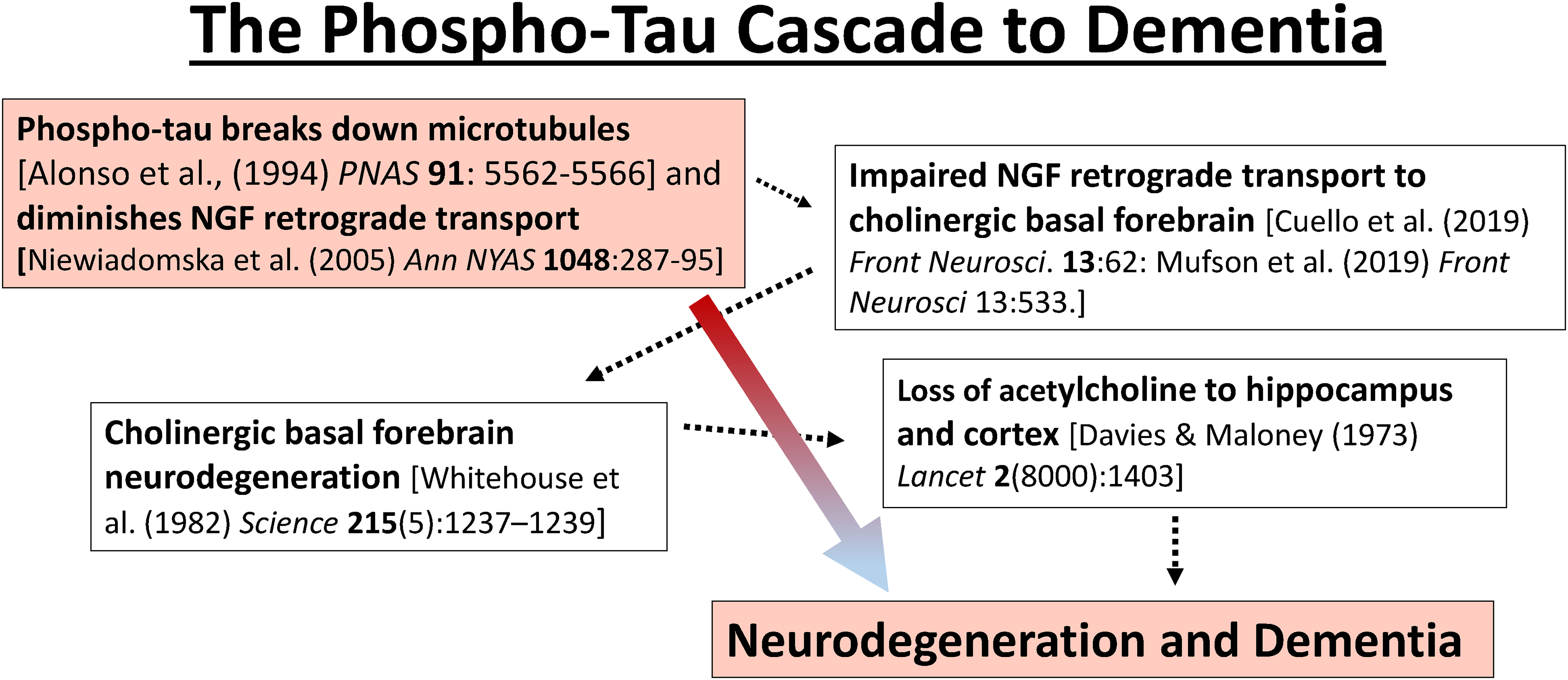
The phospho-tau cascade to AD-associated basal forebrain degeneration and dementia. The cascade begins with the hyperphosphorylation of tau and ends with impairment (atrophy) of the cholinergic basal forebrain and, finally, to dementia. Intermediate steps in the cascade such as diminished NGF transport are indicated with dotted arrows. Note: “hyperphosphorylation” denotes aberrant phosphorylation of tau protein at specific AD-associated sites (pseudohyperphosphorylation), 21 not stoichiometric (100%) phosphorylation.
Besides suggesting a final common pathway to dementia, Figure 1 also suggests a mechanism by which AChE inhibitors, originally expected to only provide cognitive enhancement, could slow basal forebrain neurodegeneration in patients with AD, including suspected prodromal patients.22–25 According to this hypothesis, AChE inhibitors may amplify acetylcholine-dependent release of pro-NGF and, thus, mitigate, or partially mitigate, phospho-tau-induced loss of NGF and improve cholinergic basal forebrain survival (Figure 1).16,25 The phospho-tau cascade hypothesis, therefore, suggests that AChE inhibitors have two modes of action. First, they act as a front-line enhancement of acetylcholine as a conventional short-acting treatment for dementia. 15 And secondly, they enhance NGF release and distribution and, thereby, promote long-term neuroprotection and neuronal survival. 25
NGF is key to the survival of the basal forebrain, and its cholinergic phenotype, in the healthy brain and its susceptibility to impairment in AD
As proposed originally by Appel, neurotransmitter release of neurotrophic factors is activity-dependent and is required to feedback and sustain the originating neurons, a loop that maintains functional synaptic connectivity. 26 For example, NGF and brain-derived neurotrophic factor (BDNF) provide activity-dependent neurotrophic signaling that is essential for neuronal survival and differentiation.27–30
The developmental origin of the basal forebrain cholinergic cells is within the subpalladium, a ventral telencephalic region that also gives rise to the basal forebrain GABAergic (gamma-aminobutyric acid) projection neurons. The ChAT-expressing basal forebrain neurons provide the long-range cholinergic projections to neocortex, hippocampus, olfactory bulb, and amygdala.31,32 It has been suggested that all basal forebrain projection neurons begin as GABAergic but that only ∼50% of these neurons eventually come to express ChAT. The remaining ∼50% that do not express ChAT are solely GABAergic.33,34 It is the ChAT-expressing neurons in the basal forebrain that contain the highest concentrations of NGF receptors in the central nervous system (CNS) and these are the ones that have a special vulnerablility to a loss of NGF and atrophy in AD.35–37 Unlike the projection neurons of the basal forebrain, the neurons of the brainstem cholinergic nuclei (laterodorsal tegmental and pedunculopontine tegmental nuclei) that project to the thalamus, ventral tegmental area, and substantia nigra, 32 do not contain significant NGF receptor concentrations and are relatively spared in AD. 35
The importance of activity-dependent neurotrophic factor release, including NGF, continues into adulthood where it is critical to maintaining active connections via target-dependent survival, 26 promoting neuronal plasticity, 38 and maintaining neuronal phenotype.39,40 Although NGF is not the only neurotrophic factor affected by AD, 41 it has garnered the most attention.42,43 The intimate relationship between NGF function and basal forebrain survival, and its failure in AD, is shown in Figure 2.
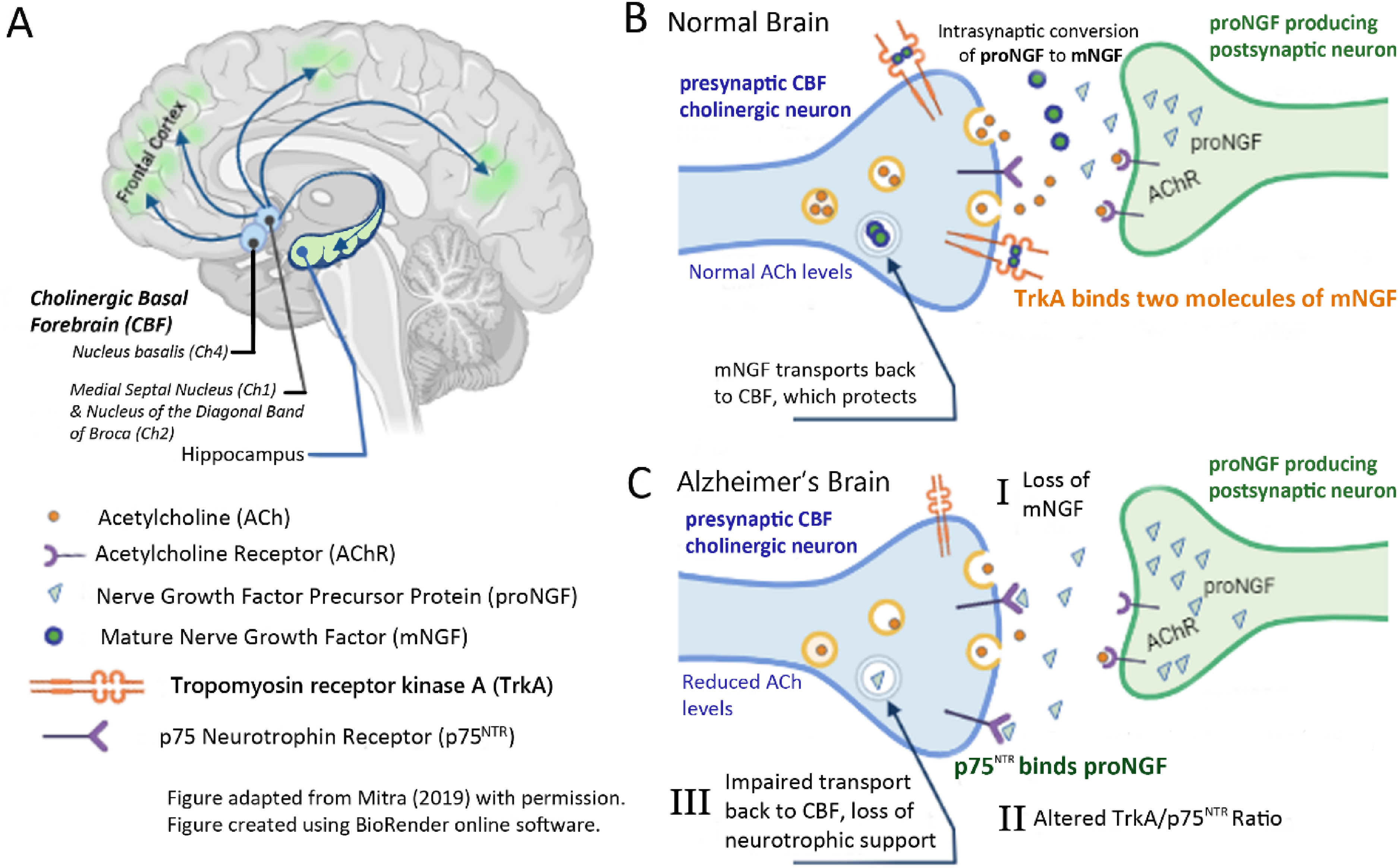
Cholinergic basal forebrain projections and NGF. (A) The cholinergic basal forebrain (CBF) and the projections of its specific nuclei to the cortex and hippocampus. (B) The source of pro-NGF and its conversion to mature NGF (mNGF), TrkA binding and reuptake of mNGF into the presynaptic CBF neuron in the normal brain. (C) Pathophysiological changes in AD which include severe impairment of NGF content and NGF retrograde transport in the cholinergic forebrain projection system and the accumulation of supernormal amounts of pro-NGF in the cortex and hippocampus.12,14,18 Acetylcholine receptor-mediated release of pro-NGF is accompanied by concurrent extracellular secretion of tissue plasminogen and neuroserpin that are mainly responsible for the intrasynaptic conversion of proNGF to mature NGF. 44 Plasminogen activated matrix metalloproteinase-9 (MMP-9) is responsible for the rapid enzymatic degradation of free mNGF that is not immediately bound to Trk-A. If not converted to mNGF or immediately destroyed, pro-NGF has a high affinity for p75NTR receptor that alters the TrkA/p75NTR ratio in favor of apoptosis.44,45 Figure adapted from Mitra et al. (2019) with kind permission. 43
The acetylcholine-NGF trophic feedback loop in the healthy brain that is required for basal forebrain survival
Figure 2 [B] shows that acetylcholine supplied by the basal forebrain axonal projections stimulates activity-dependent release of pro-NGF that is synthesized and stored in the hippocampus and cortex.44,46–50 The extracellular pro-NGF is then converted into mature NGF (mNGF) in the synaptic space, binds to TrkA receptors for endocytosis, and then undergoes retrograde transport along cholinergic axon microtubules to NGF receptors as required to sustain the viability and cholinergic phenotype of these basal forebrain neurons. 44 The cholinergic basal forebrain projections depicted in Figure 2B, therefore, fulfill two critical functions. They provide the acetylcholine that is required for short-term cognition, 51 and they stimulate the release of a target-tissue neurotrophic factor that is necessary to sustain the survival of the originating basal forebrain cholinergic neurons.26,44 Figure 3 (below) extends Figure 2B to show the basal forebrain origin of the cholinergic projections that supply the activity-dependent release of pro-NGF and the normal retrograde transport of NGF needed to sustain the basal forebrain neurons and their cholinergic phenotype. Figure 2C shows what happens during pathogenesis in AD brain.
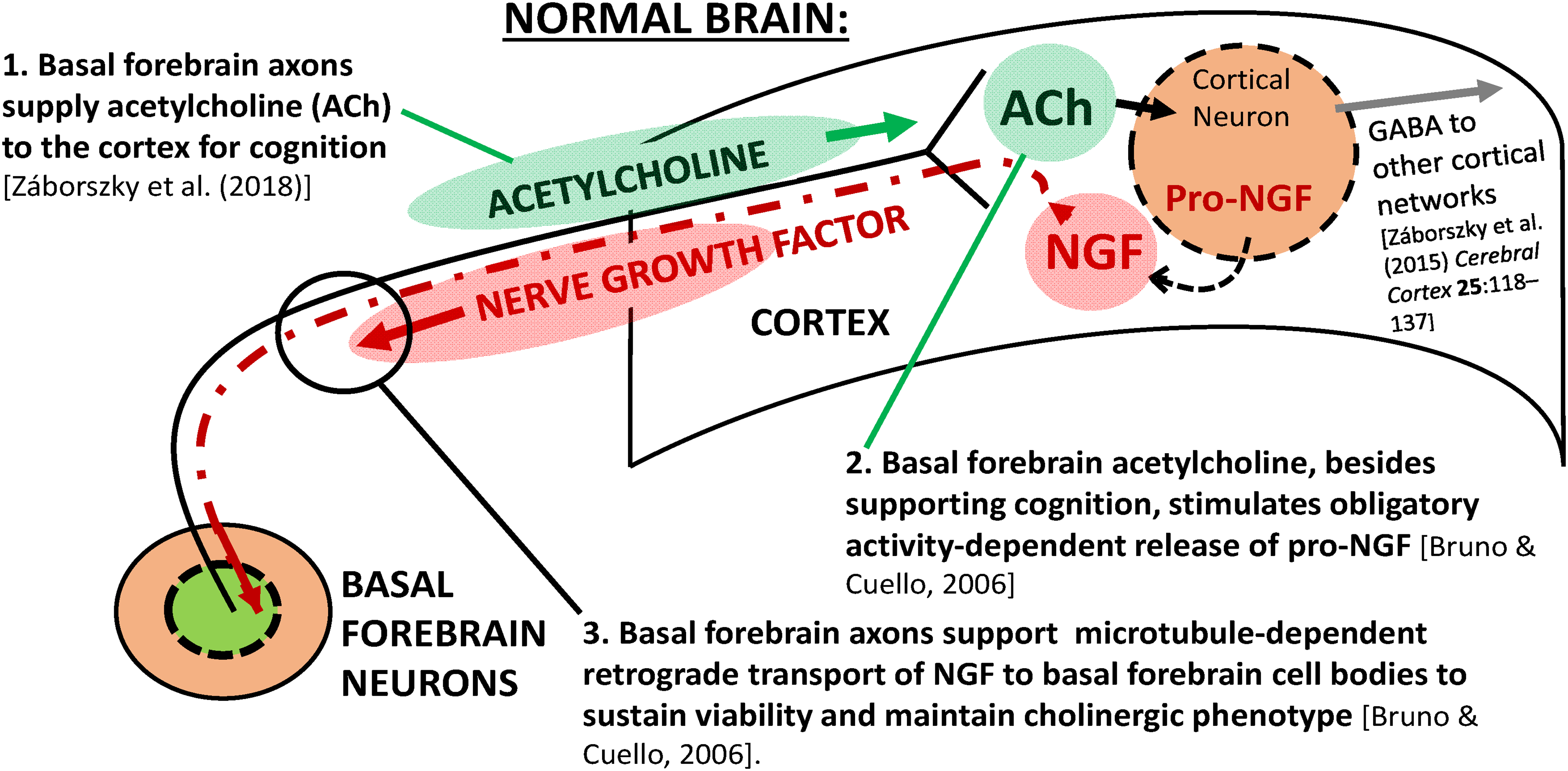
Schematic illustration of the relationship between acetylcholine from basal forebrain neurons and the cortex (green). Basal forebrain axonal projections provide cholinergic stimulation as required for functional cognition 51 (step #1) and to supply the obligatory activity-dependent activity release of cortical pro-NGF 44 (step #2). Lastly, pro-NGF is converted into mature NGF in the synaptic space, bound to TrkA receptors for endocytosis, and then undergoes retrograde transport along cholinergic axon microtubules to NGF receptors (red) as required to sustain basal forebrain neuronal viability and cholinergic phenotype. 44
Disruption of the acetylcholine-NGF feedback loop caused by phospho-tau interference with retrograde NGF transport
Figure 4 (below) extends Figure 2C to show additional details of phospho-tau-induced changes during AD impairment. The pivotal role of phospho-tau in the proposed hypothesis is its interference with microtubule-dependent retrograde transport of NGF (Figure 4). Phospho-tau is the result of pre-tangle tau hyperphosphorylation 52 and it is coincident with NGF transport dysfunction53,54 as well as the onset of both mild cognitive impairment and advancing AD. 14 AD-associated phospho-tau, the product of post-translational hyperphosphorylation, blocks microtubule-dependent retrograde transport of NGF,55–57 and causes atrophy of the basal forebrain14,58 and the loss of its cholinergic phenotype. 44
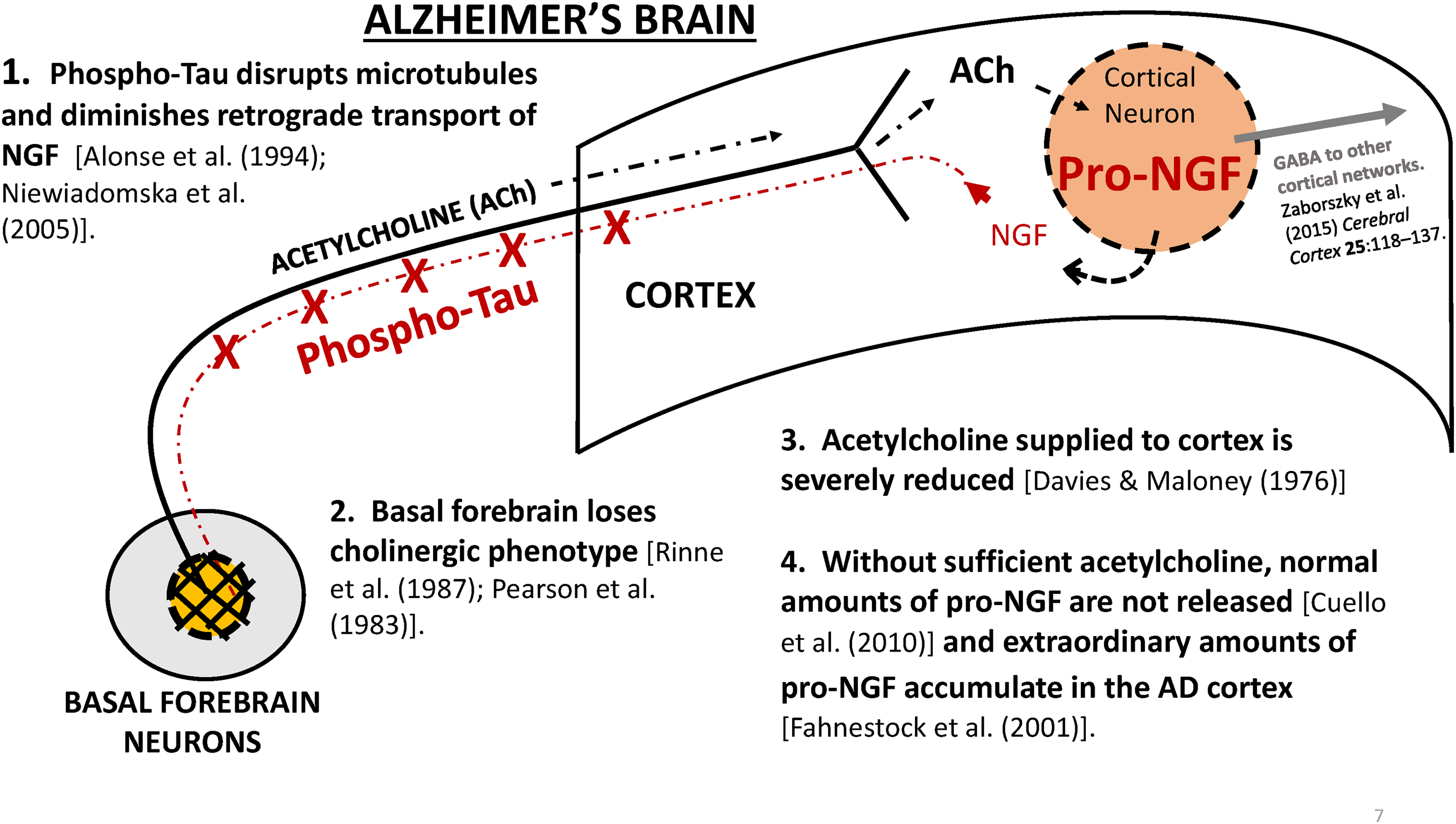
An illustration of the proposed sequence of events in the phospho-tau cascade that leads to basal forebrain atrophy and loss of cholinergic phenotype and dementia. According to this hypothesis, the cascade is initiated by the hyperphosphorylation of tau protein that interferes with the retrograde transport of NGF (Step 1, above). In the face of reduced NGF, the basal forebrain neurons atrophy and lose their cholinergic phenotype (Step 2, above). The result is that acetylcholine available to the cortex is reduced (Step 3, above). Lastly, less acetylcholine availability results in reduced activity-dependent stimulation of pro-NGF release, resulting in basal forebrain degeneration, as well as insufficient acetylcholine for moment-to-moment cognitive process, causing dementia.
The events shown in Figure 4 are supported by several findings. Besides the deficit of NGF protein itself, there is impaired NGF-dependent TrkA and p75NTR retrograde signaling12,18,59 that results in a paradoxical accumulation of pro-NGF in the cortex during mild cognitive impairment and the earliest stages of AD.60,61 The next step in the proposed cascade is that impaired NGF signaling causes presynaptic dysfunction in cholinergic neurons. 12 As the disease progresses, the deficits in neurotrophic support lead to a decline in markers for cholinergic neurotransmission that occur in the later stages of the disease.62–64 Finally, there is the appearance of dementia, the most catastrophic outward symptom. 15
The role of phospho-tau as a disruptor of microtubule retrograde transport of a neurotrophic factor, essential for neuronal survival in AD (Figure 4), is parallel to what occurs in other neurodegenerative diseases. For example, in Huntington's disease (HD) an aberrant form of the microtubule-associated protein, huntingtin, interferes with the retrograde transport of BDNF that subsequently causes the striatal atrophy that is characteristic of that disease. 30 The HD-associated mutant huntingtin protein not only fails to carry the retrograde transport of BDNF, but it also actively interferes with BDNF retrograde transport by the remaining normal huntingtin. 30 In both of these major neurodegenerative diseases, AD and HD, a dysfunctional microtubule-associated protein that is essential for the retrograde transport of a neurotrophic factor is the cause of the disease-specific pattern of striatal atrophy in HD or neurodegeneration of the cholinergic basal forebrain in AD.
Anti-neurodegenerative benefits of AChE inhibitors
The phospho-tau hypothesis and its proposed interference with NGF-dependent survival of the basal forebrain (Figure 4) provides a rational basis for the reports of AChE inhibitor-induced slowing of AD-associated neurodegeneration of the basal forebrain, hippocampus, and cortex.22–25 The hypothesized mechanism is that AChE inhibitors stimulate increased acetylcholine-dependent pro-NGF release, thereby mobilizing AD-associated pre-existing excessive pro-NGF that accumulates in the cortex during AD.41,61,65
Pro-NGF is readily released from target tissues under in vivo conditions by cholinergic receptor-mediated stimulation. 44 Insofar as the main action of AChE inhibitors is to increase acetylcholine in the synaptic space and to prolong its action on its receptors, it is proposed that AChE inhibitors, by acting as indirect agonists on acetylcholine receptors, increase activity-dependent in vivo delivery of endogenous pro-NGF directly into the synaptic space (Figure 5).
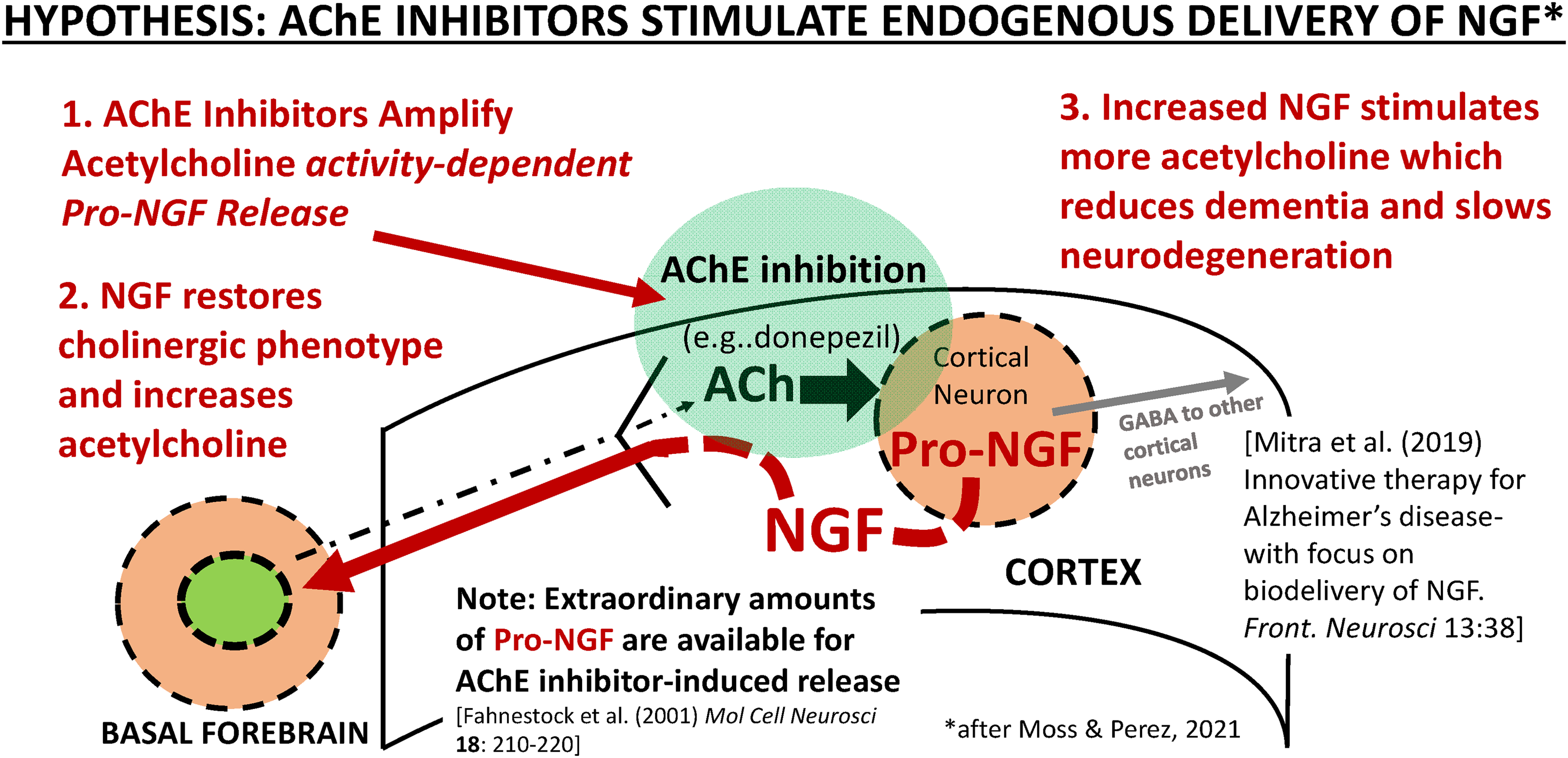
A schematic illustration of the mechanism by which anti-neurodegenerative benefits of an AChE inhibitor might be produced in AD. The key cholinergic synapse that provides the activity-dependent stimulation of pro-NGF release is highlighted in green. It is hypothesized that an AChE inhibitor (e.g., donepezil 24 ) stimulates activity-dependent release of pro-NGF. 44 Increasing pro-NGF is expected to improve NGF delivery to the basal forebrain and slow the rate of basal forebrain atrophy and restore cholinergic phenotype.25,44 In this model, AChE inhibitors achieve acetylcholine receptor-stimulated delivery of target-derived endogenous pro-NGF from the cortex and hippocampus for its conversion to NGF in the synaptic space, 44 activation of TrkA receptors, and endosome formation for retrograde transport,66,72 a goal for NGF delivery. 43 AChE inhibitor-induced release of pro-NGF does not require enhanced pro-NGF synthesis because of the pro-NGF that accumulates to paradoxically high levels in AD. 65 Modified from Figure 1, Moss and Perez, 2021. 25
Newly released pro-NFG is rapidly converted to mature NGF in the synaptic space by the co-release of the protease cascade and its endogenous regulators. 44 The mature NGF is then either internalized by TrkA activation and binding, 66 or destroyed by matrix metalloproteinase 9. 44 Pro-NGF, if not converted to mature NGF, can bind to p75NTR and increase the risk of apoptosis (Figure 2). As reviewed by others, low-affinity p75NTR receptors are co-expressed with high-affinity Trk neurotrophic receptors, but the apoptotic signals from p75NTR activation are suppressed by activated Trk receptors, biasing the outcome toward cell survival. 67
The pro-survival response to NGF requires binding to the extracellular domain and activation of TrkA receptors, cathrin-mediated macroendocytotis and retrograde propagation,66,68,69 actions that induce the formation of signaling endosomes that are internalized and propagated to the neuronal cell body by intact microtubules. 70 NGF maintains the activation of the TrkA receptors during retrograde transport to the cell body where activated TrkA and NGF then regulate neuronal survival and differentiation.66,71,72 NGF by itself is not sufficient to promote cell survival. For example, NGF injected directly into the cell body does not act directly at cytoplasmic or nuclear sites, for example to induce ChAT, and NGF antibodies do not block cell survival produced by retrogradely activated TrkA receptor/NGF signaling endosomes. 73 It has been argued that activated TrkA in endocytotic vesicles is the retrograde signal. 66 Other mechanisms of long distance NGF retrograde transport are reviewed by others.74,75
Regardless of the nature of the NGF/TrkA signaling and the mechanisms of its propagation, our hypothesis is that AChE inhibitor-induced anti-neurodegenerative benefits may be triggered by acetylcholine receptor-mediated release of pro-NGF. The subsequent long-distance retrograde propagation of signaling endosomes containing NGF and activated TrkA along intact microtubules is then required to produce the AChE inhibitor-induced anti-neurodegenerative effects (Figure 5).16,25
Figure 5 suggests that optimized AChE inhibitor therapy will require the use of CNS-selective inhibitors that can produce a higher impact on pro-NGF release than is currently available. 25 Peripheral toxicity, especially gastrointestinal adverse events, limits current AChE inhibition in the CNS to barely marginal efficacy.76–82 Secondly, in accordance with the expectation that maintaining cholinergic function will be more successful than trying restore function after it is lost,25,52 AChE inhibitor therapy should be initiated at the earliest possible time during the phospho-tau cascade, perhaps years before the appearance of cognitive decline.7,9,10
Discussion
The phospho-tau cascade raises three questions to guide future research: 1) to what degree could maximized CNS-selective AChE inhibitors enhance pro-NGF release and subsequent mature NGF production as measured in vivo; 2) if so, to what degree does enhanced AChE inhibitor-induced endogenous NGF result in the preservation of the cholinergic phenotype of basal forebrain neurons; and, 3) is it possible for a cholinergic phenotype, once lost during advancing AD, to be restored?
Whether or not a lost cholinergic phenotype can be restored by any type of NGF therapy, even with advanced biodelivery, 43 is an issue that deserves further study. However, the phospho-tau cascade hypothesis, if correct, provides a framework for preserving the basal forebrain and its phenotype, before irreversible damage has occurred. This hypothesis highlights the importance of initiating AChE inhibitors prophylactically, starting at the first accumulation of phospho-tau that may occur years before the onset of dementia. 25 It is also critical that more effective CNS-selective inhibitors be implemented to improve preservation of the basal forebrain and postpone dementia and/or slow its progression. 83 For example, it has been estimated that if a hypothetical drug could slow the onset of dementia by 5 years, the total number of patients suffering with AD could be cut by about half, 84 or if it could, instead, slow the rate of dementia progression sufficiently, the number suffering with severe dementia could be reduced by up to 80%, 84 As a practical matter, it has already been shown in several studies that long-term AChE inhibitor therapy has the capacity to slow the rate of progression of both atrophy of the basal forebrain and its projection areas, and the end product of dementia.22–24 Thus, it is highly likely that using improved AChE inhibitors (CNS-selective) prophylactically, whether or not acting by enhancing NGF as suggested here, could both delay dementia onset and progression. 25 AChE inhibitor therapy, while not addressing the origin of AD-associated amyloid-β or tau hyperphosphorylation, might reduce its impacts on suffering and economic costs.
The phospho-tau hypothesis presented here suggests that the hyperphosphorylation of tau protein is central to dementia in AD. This proposal is supported by the recent finding the neocortical tau deposition, especially phospho-tau217,85,86 is a strong predictor, perhaps superior to baseline amyloid positron emission tomography, of long-term cognitive and functional decline in a population of cognitively normal individuals. 87 The phospho-tau hypothesis provides a rational basis and a special urgency for preventing the hyperphosphorylation of tau protein at the earliest stages of the disease, before irreversible damage has occurred, as a worthwhile strategy for avoiding the catastrophic impact of AD in an aging society.88,89
Footnotes
Acknowledgments
![]() is adapted from Mitra, Behbahani, and Eriksdotter (2019) Innovative therapy for Alzheimer's disease – with focus on biodelivery of NGF. Frontiers in Neuroscience 13(38) with kind permission. doi:10.3389/fnins.2019.00038. The bibliographic assistance of Mr Eduardo Oropeza and the University of Texas at El Paso Libraries is gratefully acknowledged.
is adapted from Mitra, Behbahani, and Eriksdotter (2019) Innovative therapy for Alzheimer's disease – with focus on biodelivery of NGF. Frontiers in Neuroscience 13(38) with kind permission. doi:10.3389/fnins.2019.00038. The bibliographic assistance of Mr Eduardo Oropeza and the University of Texas at El Paso Libraries is gratefully acknowledged.
Author contributions/CRediT
Donald Moss (Conceptualization; Writing – original draft; Writing – review & editing); Ruth G. Perez (Conceptualization; Writing – original draft; Writing – review & editing).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.