
Abstract
Keywords
Introduction
The zoonotic orthopoxvirus, a member of the poxviridae family causes monkeypox. 1 Animals like rodents and monkeys are the primary carriers, but humans are also involved. 2 In 1958, the virus was found in monkey’s body in a laboratory in Copenhagen, Denmark. 3 As a result of an intensified campaign to eradicate smallpox, the Democratic Republic of the Congo reported the first case of monkeypox in 1970. 4 However, monkeypox is mostly dominant in central and western regions of Africa, where people who live near tropical rainforests. 5 In West and Central Africa, only 50 cases of monkeypox were reported in 1990. In 2020, there were more than 5000 cases all over the world, whereas in the past, monkeypox was thought to exist only in Africa. In the recent year, 2022, it was reported in several of non-African countries, including the United States and Europe. 6 The Centers for Disease Control and Prevention (CDC) estimated that as of August 23, 2023, there were approximately 89,385 monkeypox patients globally, and 94 nations reported cases of monkeypox in 2022. 7 As a result, people are gradually becoming more and more anxious and afraid, which is frequently reflected in people’s opinions on social media, 8 as there is currently no effective treatment for the monkeypox virus, according to the CDC’s recommendations.
The rapid gathering of information gathering is needed to develop an effective therapeutic treatment for this infectious disease. The Next Generation Sequencing (NGS) is a widely used way to gather enormous volumes of data in a relatively quickly. When many more organisms are sequenced, the challenge of assigning functions to genes is increasing9,10. These genes have translated into multiple types of crucial proteins, and more than 30% of their molecular activities are unknown. These are termed as Hypothetical Protein (HP). 11 The monkeypox virus contains many hypothetical proteins whose functions are unknown. Proper treatment might be generated by knowing the structural and functional characteristics of a hypothetical protein of the monkeypox virus. Furthermore, structural and functional annotation of HPs may also reveal potential biomarkers and pharmacological targets. 12 With the use of many updated algorithms and software, bioinformatics tools give a platform for determining the structure and function of hypothetical proteins through homology modeling or domain homology searches. 13 Thus, the objectives of the present study are to use bioinformatic tools based on various algorithms to annotate the structural and functional fractures of a hypothetical protein (accession no. URK21192.1, PDB ID 6zyc.1). Based on the findings of the study, the hypothetical protein played a key role in a complete scenario of viral replication and host-pathogen interaction. Nevertheless, this study may provide insight into a new approach to the design and discovery of drugs.
Materials and methods
Sequence retrieval
The NCBI protein database was searched for the HP (accession no. URK21192.1) of the monkeypox virus, which led to of the amino acid sequence selection. The sequence’s FASTA format was retrieved and submitted to several prediction servers for the in-silico characterization.
Physiochemical properties analysis
The physical and chemical properties of the target protein sequence including molecular weight, amino acid composition, theoretical pI, instability index, extinction coefficient, atomic composition, estimated half-life, the total number of positively charged residues (Arg + Lys), the total number of negatively charged residues (Asp + Glu), aliphatic index, and grand average of hydropathicity (GRAVY) were analyzed using ExPASy’s ProtParam tool. 14
Identification of conserved domains, motifs, and super families
Conserved Domain Database (CDD, available at NCBI), 15 Pfam, 16 and InterProScan 17 were used for domain analysis. The Motif 18 server was used to find protein motifs. The protein folding pattern was identified using the PFP-FunDSeqE.
Multiple sequences alignment and phylogeny analysis
BlastP 19 from NCBI was used to find similarities in the non-redundant (nr) sequences. At the same time, multiple sequence alignment and phylogeny analyses were carried out by using CLC sequencer viewer 8.0. In this case, the neighbor-joining method for constructing the tree was implemented, and an unrooted phylogeny model was portrayed.
Prediction of secondary structure
The self-optimized prediction method with alignment (SOPMA) was used to predict the secondary structure of the target protein. 20 To ensure the accuracy of SOPMA results, PSIPRED 21 was also implemented. Furthermore, assessment of the model was assessed to check its validity and structural conformation.
Three-dimensional (3D) structure prediction
The SWISS-MODEL server 22 was used to determine the 3D structure of the target protein based on homology modeling. The server automatically performs a BLASTp search for potential templates for each protein sequence. Template protein 6zyc.1. A was chosen for homology modeling from the search result with 96.75% sequence identity, which was a reliable score to start modeling; this is an X-ray diffraction model of C-terminal domain of the vaccinia virus DNA polymerase processivity factor component A20 protein
Energy minimization of the 3D model
The overall energy of the 3D structure from the SWISS-MODEL server was minimized using the YASARA force field minimizer. 23 Comparatively lower energy, precise 3D structure provides stable configuration of the target protein. Therefore, a high throughput field of the YASARA program was utilized to reach optimum energy stability because stable configuration should not be compromised to get additional information.
Model quality assessment
The PROCHECK, 24 Verify3D, 25 and QMEAN 26 programs of the ExPASy server of SWISS-MODEL Workspace and ERRAT 27 were used to analyze the quality of the three-dimensional structure. The ProSA server estimated the Z-score for the template and the HP. 28 Finally, the aforementioned programs were utilized to check the behavior of the protein verifying the structural assortment and overall quality.
Active site detection
The protein’s active site was identified using the computed atlas of surface topography of proteins (CASTp) server. 29 The CASTp provides precise, thorough, and quantitative information on a protein’s topographical characteristics. It is possible to precisely find and measure active pockets on protein surfaces and the interior side of three-dimensional structures. As a result, it has evolved into a platform essential for the predicting of the protein regions that interact with ligands. Additionally, FTsite was used for high-accuracy detection of ligand binding sites. It is an accessible a free web-based server at https://ftsite.bu.edu.
Molecular docking analysis
Docking analysis was performed using PyRX software. It is used to observe how the ligands are binding with the protein. The ligands used for the docking were tecovirimat (TPOXX) CID_ 16124688, cidofovir (CID_ 60613), and ribavirin (CID_ 100252). The binding affinity, 3D, 2D structure, and docking results were observed and analyzed by BIOVIA Discovery Studio software.
Analysis of molecular dynamics simulation
Schrodinger Maestro version 11.8 generated simulation snapshots depicting individual atomic movements. The simulation data was then analyzed using the Simulation Interaction Diagram (SID) feature provided within the Schrodinger package. In addition to the root mean square deviation (RMSD), metrics such as root mean square fluctuations (RMSFs), radius of gyration (Rg), and solvent accessible surface area (SASA) values were also investigated.
Results
Physicochemical properties
Physicochemical properties of the MPXV-SI-2022V502225_00135 estimated by ProtParam tool.

Function prediction by domain and motif analysis
Functional annotation results of different tools.

Multiple sequences alignment and phylogenetic tree
Non-redundant sequencing information of proteins with similar properties.


Multiple sequence alignment (MSA) among different DNA polymerase processivity factor protein family of monkey pox using CLC Sequence Viewer version 8.
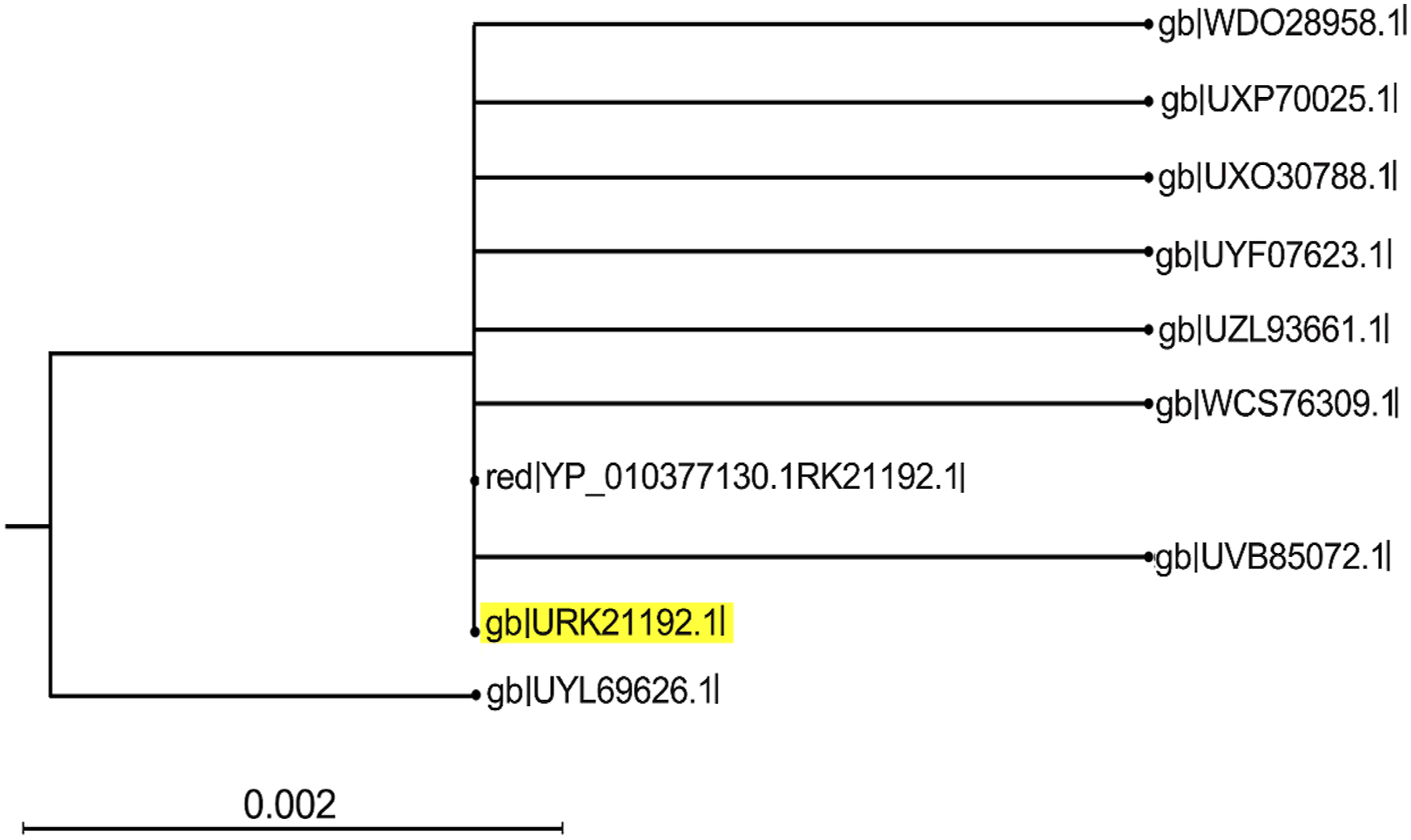
Phylogenetic tree showing evolutionary relationship of the target protein with DNA polymerase processivity factor proteins.
Prediction of secondary structure
In this case, the secondary structure of the protein was predicted by PSIPRED, SOPMA, and ENDscriptserver. According to SOPMA estimation, the alpha helix was found to be the most predominant (42.49%), followed by extended strand (22.77%), random coil (27.46%), and beta-turn (7.28%). Similar results were obtained from ENDscript and PSIPRED (alpha helix: 38.26%, random coil: 34.50%, and extended strand: 26.99%). The secondary structure of the protein predicted by PSIPRED is shown in Figure 3. Predicted secondary structure of the target protein using PSIPRED server.
Tertiary structure prediction and energy minimization
The HP tertiary structure was obtained from the SWISS-MODEL server using the template 6zyc.1, which shows 96.75% sequence identity with the target protein. The structure obtained through SWISS-MODEL is depicted in Figure 4. The YASARA Energy Minimization Server minimized the energy of the modeled protein from −52684.8 KJ/mol to −72589.0 kJ/mol. The preliminary score was −1.77, but after energy minimization, the final score turned to 0.39, indicating a more stable form. Predicted 3-dimensional structure of the target protein through SWISS-MODEL server after YASARA energy minimization.
Model quality assessment
Ramachandran plot analysis.

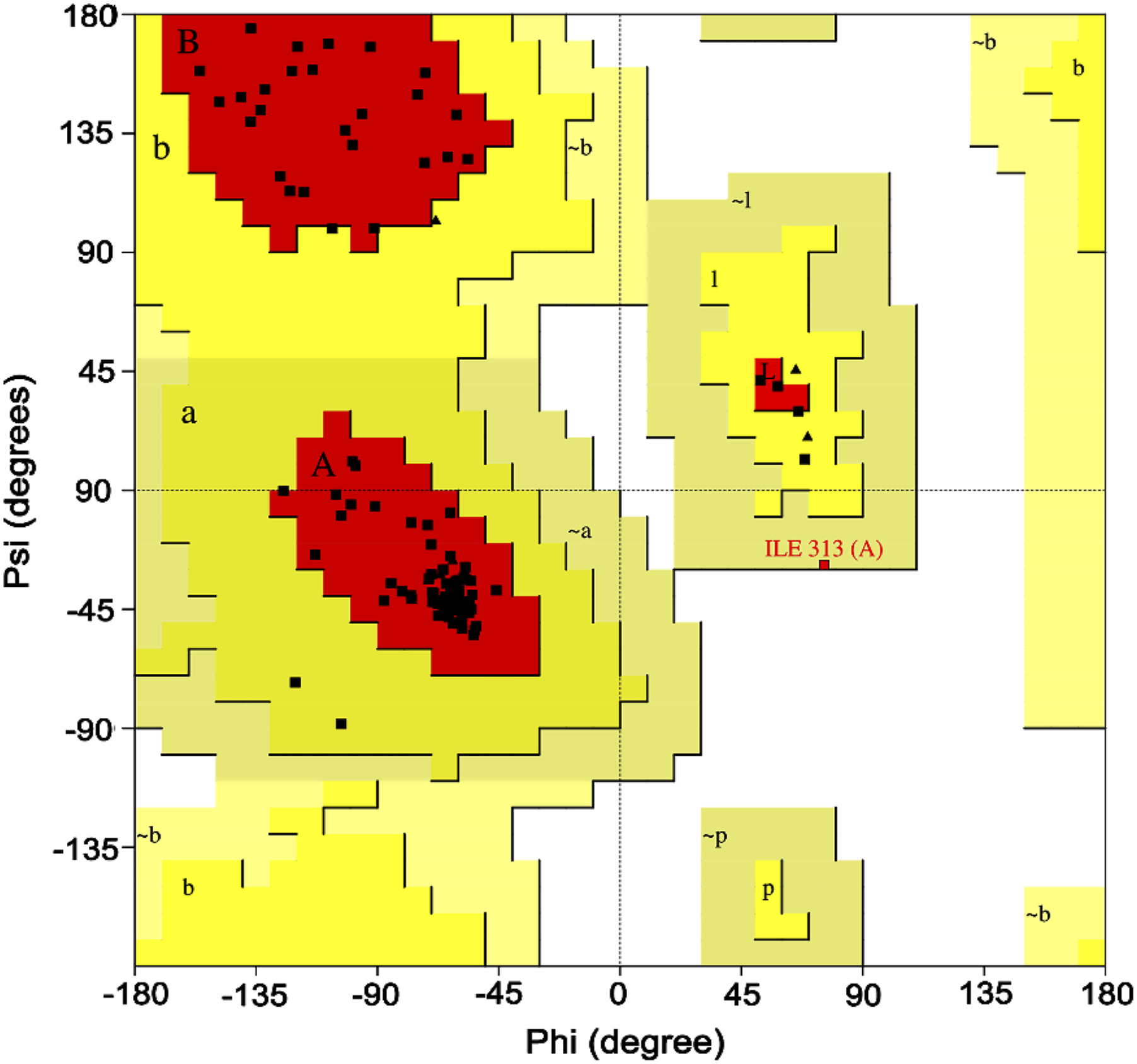
Quality assessment of the model of Ramachandran plot of model structure validated by PROCHECK program.

Graphical representation of QMEAN result (a), Z-score (b), and Verify3D result (c) of the model structure.
Active site determination and molecular docking analysis
The active site was predicted by using the CASTp server (Figure 7). CASTp is a database server that can recognize regions on proteins, determine their boundaries, compute the area of the areas, and calculate the dimensions of the areas. Vacuums concealed within proteins and pockets on protein surfaces are also involved. To define a pocket and volume spectrum or vacuum, surfaces of solvent accessible molecules (Richard surface) and molecular surfaces (Connolly surface) are employed. However, the most active site was predicted in to be one of the largest enormous pockets, having 39.371 solvent accessible (SA) surface area and a total volume of 13.707 amino acids. Figure 7(a) displays the key active residues predicted from pockets are LYS306, TYR307, PHE 308, SER 309, GLU 380, LYS 383, VAL 385, and ASN 415. Moreover, the ligand binding sites were detected and observed by the web-based server FTsite. This binding site shows the functional relationship between protein-ligands. Active site predicted by CASTp server representing (a) the active location of HP in red sphere and active amino acid residues are highlighted in gray color also and (b) the active site chain A.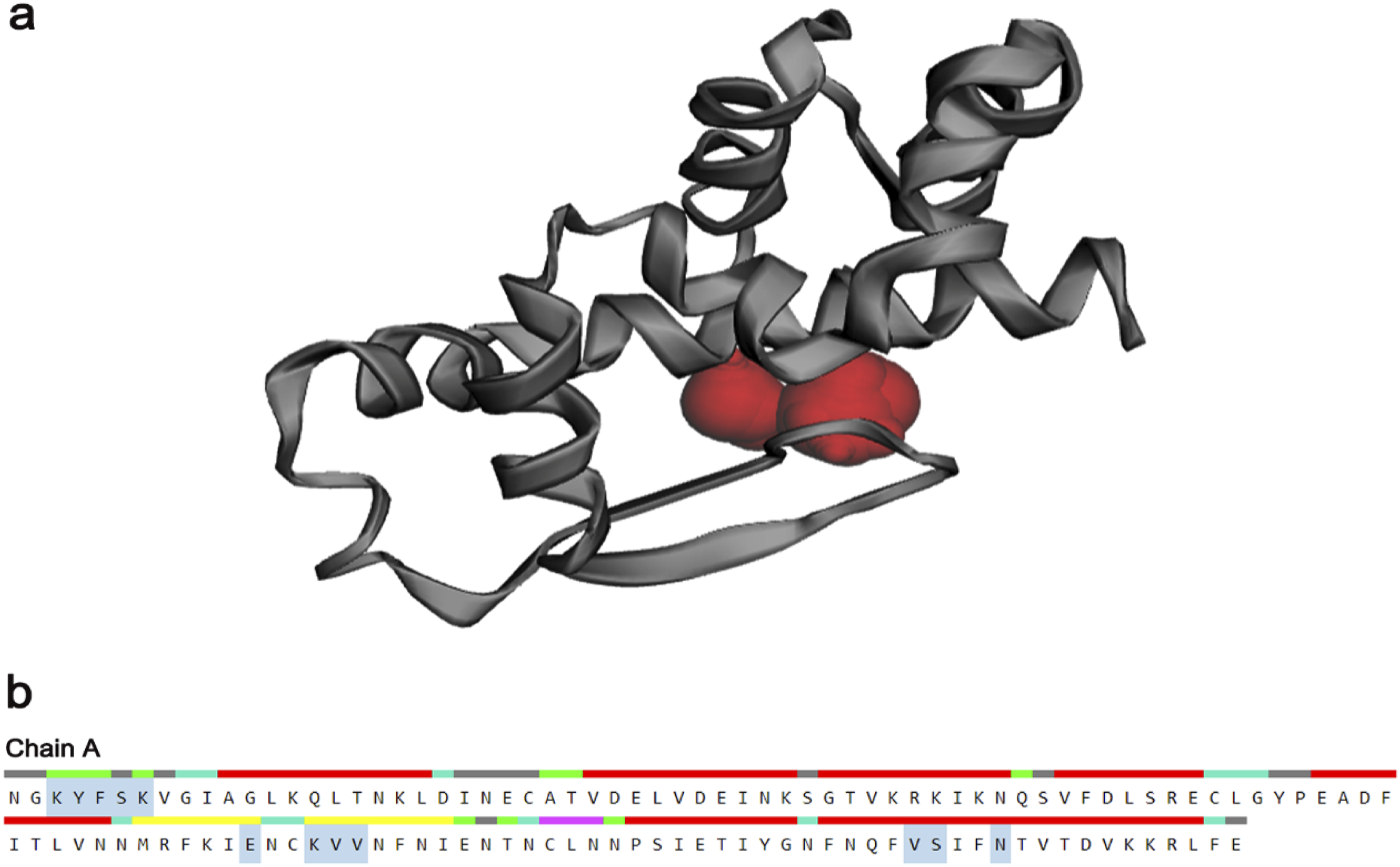
Binding interaction of tecovirimat (CID_ 16124688) with hypothetical protein (URK21192.1).

The docking score of different ligands with hypothetical protein.


(a) Predicted three-dimensional structure of the target protein binding with the ligand visualized by BIOVIA Discovery Studio. It shows a ligand (CID_16124688) bound with hypothetical protein URK21192.1 and (b) 2D structure of protein-ligand binding complexes.
Analysis of molecular dynamics
Molecular dynamics (MD) simulation plays a crucial role in post-dock analysis, enabling the exploration of time-dependent stability and atom movements within the biological environment. Essential analyses within MD simulations include RMSD, RMSF, SASA, and RG, collectively offering a comprehensive grasp of the molecular behavior of protein-ligand complexes. These analyses were conducted following a 100 ns dynamics trajectory for the monkeypox protein 6zyc.1 and the ligand tecovirimat (CID_ 16124688).
RMSD analysis
RMSF stands for root mean square fluctuation. Root means square fluctuations (RMSFs) determine the individual amino acids in a complex system. It calculates individual residue flexibility or how much a particular residue moves (fluctuates) during a simulation.
30
Based on the atomic root mean square deviation (RMSD) of protein 6zyc.1 backbone, we can evaluate the change in displacement of selected atoms during a period.
31
The RMSD (Figure 9(a)) shows that the value of Tecovirimat with the 6 zyc.1 from 0 to 50 ns was stable, but after that, it was less stable. A sudden fluctuation was seen at 30ns of 0.534 Å. The maximum, minimum, and average RMSDs of 6zyc.1 are 0.855 Å, 0.126 Å, and 0.421 Å. Illustrating that RMSD, RMSF, Rg, and SASA values of the complex structure extracted from 6yzc: CID_16124688 concerning 100 ns simulation time.
RMSF analysis
RMSF stands for root mean square fluctuation. RMSFs determine the individual amino acids present in a complex system. It calculates of individual residue flexibility or how much a particular residue moves (fluctuates) during a simulation. 30 It showed continuous minor fluctuations around 3 Å during simulation (Figure 9(b)).
Radius of gyration (Rg)
The radius of gyration is the measurement of the distance of the root, which means square radial, from the center of mass of the target protein to both of its terminals. By this measurement the compactness, of the protein-ligand complex can be understood from the value of protein mobility and rigidity with the corresponding ligands. 32 The protein-ligand complex was stable to the whole of the 100 ns (Figure 9(c)). The minimum, maximum, and average Rg values of tecovirimat (CID_ 16124688) are, respectively, 4.984 Å, 4.689 Å, and 4.86 Å which demonstrate higher mobility and rigidity of 6 zyc.1 with the compound.
Solvent accessible surface area (SASA)
The solvent accessible surfaces of protein-ligand complexes explain their solvent-like behavior (hydrophobic or hydrophilic). 32 Each complex was plotted based on its surface area, which is assessable to solvent molecules (SASA) in (Figure 9(d)). The figure demonstrates good tight bonding of the compounds with the hypothetical protein.
Discussion
Monkeypox (MPX) is a zoonotic disease, but its animal reservoir remains unknown. Various rodent species from Central and West African tropical rainforests, including tree squirrels and Gambian pouched rats, are currently considered to be strong candidates.33,34 However, monkeypox viruses belong to the family of Poxviridae, a subfamily of Chordopoxviridae, and the genus of orthopoxvirus. 35 There are two genetic clades, with genomes differing by less than 1%. The first clade is endemic in Central Africa, and the second in West Africa.36,37 Various symptoms of MPX include fever, head and muscle aches, lymphadenopathy, and a characteristic rash that develops into papules, vesicles, and pustules, which eventually scab over and heal. Unfortunately, there are no licensed vaccines available for MPX, but researchers are striving to develop a Monkeypox vaccine. Therefore, over 70% of people living today are never vaccinated against smallpox. 38
Research on hypothetical proteins has yet to keep up with the rapid development of low-cost sequencing technology despite the enormous amount of genomic and proteomic data that has been generated. 39 Characterization of hypothetical proteins can improve the knowledge of viral metabolic pathways, disease progression, drug development, and disease control strategies. 40 In this study, we analyzed physicochemical properties, and the protein was estimated to contain 426 amino acids with a molecular weight of 49147.14, theoretical pI 5.62, aliphatic index of 92.77, grand average of hydropathicity (GRAVY) of −0.258, and the instability index of the target protein 36.35 (Table 1).
In this investigation, domain and motif analysis predicted our target hypothetical protein to be a Chordopox_A20R domain-containing protein by all the annotation tools with high-level confidence (Table 2). The BLASTp results against the non-redundant (nr) database showed homology (above 99% sequence similarity) with other known A22 R (Monkeypox virus Zaire-96-I-16) from different Monkeypox virus Zaire-96-I-16, validating the prediction (Table 3).
There are several Chordopoxvirus A20R proteins in this family. The viral proteins encoded by the D4R, D5R, and H5R open reading frames directly interact with the A20R protein, contributing to assembly or stability of the multiprotein DNA replication complex. 30 Furthermore, it was estimated that the viral protein has a necessary mode of action for DNA replication and is linked with the processive form of the viral DNA polymerase. A20R may influence the multiprotein DNA replication complex’s stability or ability to assemble. Interestingly, D4R, D5R, and H5R encode uracil DNA glycosidase, nucloeside-triphosphatase (NTPase), and late transcription factor VLTF-4, respectively. It was also suggested that unlike VLTF-1, -2, and -3, that which were synthesized with elevated levels after viral DNA replication, VLTF-4 is synthesized both before and after viral DNA synthesis. However, observing its expression pattern and subcellular distribution, it was reported that the H5R gene product may have different role for a productive infection throughout the viral life cycle. 41 Uracil DNA glycosidase in the poxviral protein is responsible for the repairing of the viral DNA genome, involving removing uracil from DNA to crate apyrimidinic site. 42
The secondary structure includes random coils, alpha helices, beta-turns, and extended strands, with the alpha helices being the predominant form. The protein’s three-dimensional (3D) structure obtained using the SWISS-MODEL server successfully passed all model quality assessment tools, such as PROCHECK, Verify 3D QMEAN, and ERRAT. The 3D structure became more stable after YASARA energy minimization was 0.39. Accordingly, the Z-score was calculated −6.0, indicating the overall model quality is good (Figure 6(a)–(c)). Local model quality was observed as a stable profile in terms of the regarding knowledge-based energy of the residues.
The protein’s folding arrangement creates a pocket or groove known as the active site. However, the active amino acid residues were computed by CASTp server and were consistent with the prediction of functional annotation tools and lie in the Chordopoxvirus A20Rsuperfamily domain region. Its three-dimensional structure, as well as and the electrical and chemical characteristics of the co-factors and amino acids in the active site. The result of the investigation reported that the active site had a solvent accessible surface area (SASA) of 39.371 and the amino acids volume of 13.707 (Figure 7). Molecular docking was performed by PyRx 0.9 software to know the interaction among the target protein and ligands. These ligands are, respectively, tecovirimat, cidofovir, and ribavirin. A strong solid binding affinity was found for the hypothetical protein and tecovirimat. Their docking score was comparatively very well. It was about −7.1. The 3D and 2D structures with protein-ligand (HP and tecovirimat) complex are placed in Figure 8. Many binding interactions of tecovirimat (CID_ 16124688) with hypothetical protein (URK21192.1) are presented in the Table 5; the ligand binding sites were also observed by FTsite software (Figure 10). Identifying binding sites is a classic problem important significant for many applications, including protein engineering and drug design, structure-based prediction of function, and elucidating functional relationships between proteins.
43
Molecular dynamic simulation was done with the best docking result compound tecovirimat (CID_ 16124688). It showed comparatively good results, so we went for the simulation. The result shows moderate level of stability after analyzing the whole molecular dynamic simulation with this compound. The interactions with ligands were previously reported with varied compounds.44,45 Identification of binding site by FTsite after modifying by BIOVIA discovery studio.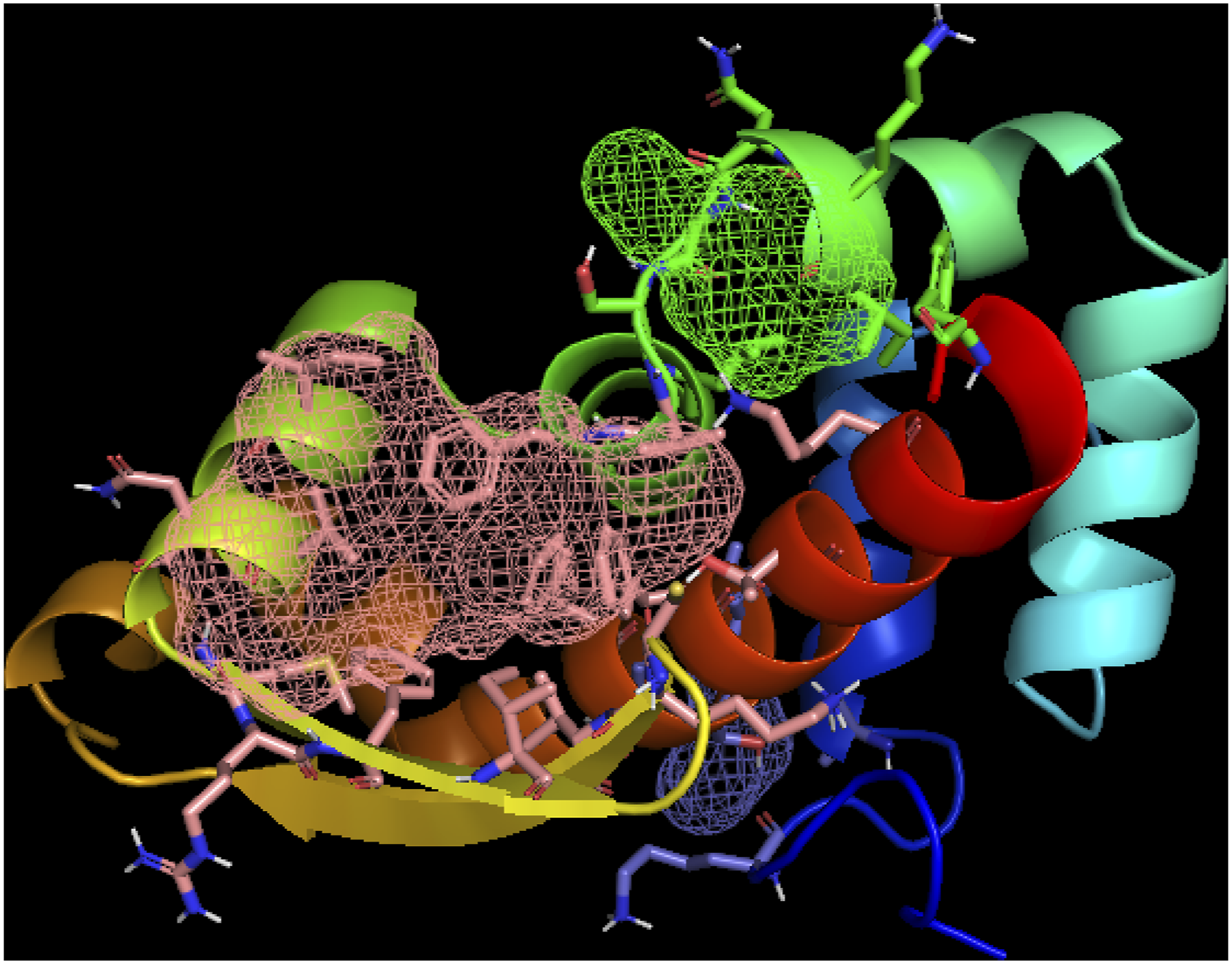
This hypothetical protein annotation strategy helps in designing of effective medicines/vaccines against MPX. Studying individual domains and/or effectors also helps to understand antiviral mechanisms. Finally, this clearly underlines the importance of continued research into Chordopoxvirus A20R and it’s their effectors not only in monkeypox but also in other pathogenic microorganisms to develop future treatment strategies.
Conclusion
This protein contains numerous characteristics in the system that are severe threat to the human health. In recent years, significant work has been done made in to understand the functions of the Chordopoxvirus A20R protein. Besides, many structural and functional aspects of this protein and its effectors are still unknown. Annotation of the hypothetical protein may help in design an effective drug or vaccine. The study will also help understand antiviral mechanisms. Further research and experimental validation are needed to confirm our findings about this crucial protein.
Footnotes
Acknowledgments
We thank to the Department of Biotechnology and Genetic Engineering, Noakhali Science and Technology University, Noakhali-3814, Bangladesh, for giving us opportunities to use the facilities to conduct the research. Moreover, we are very thankful to Md. Aktaruzzaman and the Laboratory of Pharmaceutical Technology, Department of Pharmacy, Jashore University of Science and Technology, Jashore, Bangladesh for partially providing us with computer-based support.
Author contributions
MIH and MNI performed the experiments, interpreted the data and wrote the draft manuscript. MEM, MNI, ASMF, and SHJ analyzed the data and edited the draft manuscript. MUSK, THE, MAH, FA, and MAK reviewed and edited the manuscript. SHJ formatted the figures and tables. MAM contributed to the study conceptions, design, analysis of the data, review, and editing manuscript. All the authors have approved the final version.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.