
Abstract
Background
The GSK3732394 multivalent protein was developed as a novel, long-acting, antiretroviral biologic treatment regimen with three independent, non–cross-resistant mechanisms for inhibiting HIV-1 entry.
Methods
A single-centre, Phase 1, double-blind, randomized, placebo-controlled study was conducted in healthy volunteers, using a 2-part adaptive study design: in Part 1, participants were randomized to receive subcutaneous injection of GSK3732394 or placebo (3:1) as single ascending doses (10-mg starting dose); in Part 2, participants were intended to receive multiple ascending doses. Primary and secondary objectives included safety, pharmacokinetics (PK) and pharmacodynamics (PD; cluster of differentiation four receptor occupancy [CD4 RO]) of GSK3732394 in healthy adults; PK/PD results in healthy volunteers were used to project HIV-1 treatment success.
Results
The most frequently reported adverse event was injection site reactions (ISRs; 8/18 [44%]). Most ISRs were mild (Grade 1–2; n = 7); one participant experienced a Grade 3 ISR (erythema ≥10 cm). All ISRs were delayed in onset (after Day 10). GSK3732394 demonstrated linear PK across all cohorts. Clearance was faster than expected, and PK/PD results were lower than expected, with the maximum dose investigated (80 mg) achieving mean trough CD4 RO of ∼25% on Day 7. The study was terminated as the PK/PD model linking PK and CD4 RO indicated that the maximum planned doses would not achieve the desired therapeutic profile.
Conclusions
This study demonstrated successful deployment of PK/PD dose relationships in the design and conduct of clinical trials by leveraging the findings toward predicting probability of success, resulting in appropriate early termination (ClinicalTrials.gov, NCT03984812).
Introduction
For people living with HIV, the 2020 International Antiviral Society–USA guidelines recommend combination antiretroviral therapy (ART), generally including two to three active drugs from ≥2 classes [1]. Although single-tablet formulations are available, the burden of lifelong daily dosing can contribute to imperfect adherence, potentially leading to treatment failure, emergent resistance and increased pill burden [1,2]. Daily dosing can also factor into HIV-associated stigma: individuals may skip medication to avoid reminders of disease or inadvertent disclosure [3,4]. Long-acting agents may help simplify treatment and increase adherence. To that end, cabotegravir (an integrase strand transfer inhibitor) plus rilpivirine (a non-nucleoside reverse transcriptase inhibitor) has been approved as a once- or bi-monthly, intramuscular, long-acting, injectable 2-drug regimen for virologically suppressed individuals [5].
GSK3732394 is a biologic molecule developed to offer a novel approach to HIV-1 therapy by functioning as a long-acting tri-specific entry inhibitor [6]. GSK3732394 was designed as a multivalent protein that could serve as a complete treatment regimen for viremic and/or virologically suppressed individuals with HIV-1.
GSK3732394 has three independent, non–cross-resistant mechanisms for inhibiting HIV-1 entry (Figure 1A). Two mechanisms are contributed by adnectins, single protein domains derived from the 10th type III domain of human fibronectin that bear structural similarity to the heavy chain variable domain of a typical antibody [7]. Adnectins can be altered through various efficient in vitro techniques to bind tightly to any target sequence. One adnectin in GSK3732394 binds to domains two and three of cluster of differentiation four (CD4) cell-surface receptors on human immune cells and bends CD4 toward the cell membrane such that HIV-1 can no longer bind to domain one, thus inhibiting virus binding to CD4 via steric hindrance [8].
The second adnectin in GSK3732394 binds to the N17 region of gp41 and works synergistically with the anti-CD4 adnectin, inhibiting the conformational change needed for virus–host cell fusion [9]. The third inhibitor is a helical peptide similar to the injectable antiretroviral enfuvirtide, binding to the heptad repeat one region within gp41 and working synergistically with the anti-gp41 adnectin to further inhibit the fusion step [6]. When tested separately, each individual inhibitor inhibits HIV-1 viruses with low nM activity. However, when linked together, there are multiple synergies such that GSK3732394 exhibits broad-spectrum, low-picomolar antiviral activity. This potency is unaffected when GSK3732394 is placed in human serum. Additionally, at the amino terminal end of the molecule, GSK3732394 contains a human serum albumin (HSA) domain, which functions as a pharmacokinetic (PK) enhancer, extending in vivo drug half-life [6]. The carboxy terminus of HSA is connected to the amino terminus of the anti-CD4 adnectin, which in turn is connected via a short linker to the anti-gp41 adnectin. The fusion inhibitor peptide is connected to the carboxy end of the anti-gp41 adnectin via a short linker. Adding the HSA molecule reduces potency by about 3-fold to a half-maximal effective concentration (EC50) of ∼270 pM [6].
The greatest synergy exhibited by GSK3732394 is driven by the binding of the anti-CD4 adnectin to its target [6]. Besides the intrinsic antiviral activity of this binding, it also acts as an anchor to localize the gp41-targeted components to their site of action at the cell membrane, thus synergizing antiviral potency of these molecules. This high potency was observed even at low CD4 receptor occupancy (RO), with only ∼0.2% of CD4 molecules on MT-2 cells bound with GSK3732394 at an EC50 concentration of 0.27 nM in cell culture, while ∼1.5% of CD4 molecules are bound at an EC90 concentration of 2 nM [6]. This is possibly explained by the multi-specific nature of the GSK3732394 multivalent protein and its potential ability to work in trans. While the receptor-bound anti-CD4 component blocks the initial stages of viral entry on one CD4 molecule on the T-cell surface, the anti-gp41 components of the same GSK3732394 molecule could inhibit the cell fusion step initiated by a virion attached to a different CD4 (unoccupied by GSK3732394) on the same host cell.
The therapeutic potential of GSK3732394 was assessed in an in vivo animal model. Subcutaneous administration of three different GSK3732394 doses (4, 12.5, 32 mg/kg) in an HIV-1–infected humanized mouse model yielded significant viral load reductions. At the highest dose (32 mg/kg), virologic response was commensurate with standard-of-care combination ART (tenofovir disoproxil fumarate, lamivudine and raltegravir) beyond 1 month of dosing at corresponding plasma GSK3732394 trough concentrations of 15–25 nM and mean CD4 RO rates of 40%–60% [6].
GSK3732394 was then transitioned to clinical development. After appropriate preclinical studies, an adaptive, Phase 1, first-time-in-human study was designed. Primary and secondary objectives were to characterize safety, PK and CD4 RO of GSK3732394 in healthy adults. Human dose predictions were based on PK/pharmacodynamic (PD) modelling results from the HIV-1–infected huCD4 mice study. The pre-specified PK/PD CD4 RO targets were to potentially produce robust virologic efficacy in suppressing HIV-1 with a relatively high resistance barrier and could be uniquely evaluated in healthy participants in this study. Specifically, the PK/PD targets were to achieve Ctrough concentrations necessary to provide ∼90% CD4 RO with a minimum dosing frequency of once weekly.
Methods
Study design
A single-centre, Phase 1, double-blind, randomized, placebo-controlled, single and multiple ascending dose study was conducted between June 2019 and March 2020 at a single centre in Baltimore, MD (ClinicalTrials.gov, NCT03984812; study number, 207863). The study was approved by Aspire Institutional Review Board (Santee, CA) and performed in accordance with international laws and guidelines consistent with the Declaration of Helsinki principles. Participants provided written informed consent before enrolment.
This study was planned to be conducted in two parts using an adaptive study design. In Part 1, participants were randomized to receive GSK3732394 or placebo in single ascending doses up to a maximum of four subcutaneous injections (maximum volume of 2 mL each). A starting dose of 10 mg of GSK3732394 was chosen based on preclinical data [6], minimum anticipated biological effect level and human equivalent dose analyses, and published CD4 receptor binding data for ibalizumab. At 10 mg, GSK3732394 was projected to have a maximum observed concentration (Cmax)-associated RO of ≤60% with a 7-day trough-associated RO of ≤10%. Subsequent doses were chosen to achieve predetermined RO levels unless constrained by safety-determined dose-elevation limits or the maximum dose increase of 4-fold. Six cohorts were planned to receive GSK3732394. If targeted Ctrough RO (≥90%) was not achieved by single ascending dosing in Cohort 5, then the addition of an expansion cohort (Cohort 6) was permitted. Part 1 also included sentinel dosing (1 active: 1 placebo). Transition to Part 2 with multiple ascending dosing was dependent on achieving mean Ctrough RO rate ≥20% across active participants in ≥1 single ascending dose cohort. Accumulating safety and PK/PD data were reviewed internally before each dose escalation to support proceeding to the next higher dose level and selecting the next dose. Pre-study projections suggested that ≥90% target engagement at Ctrough could be achieved in Part 1.
Participants
Investigators enrolled HIV-negative adults (aged 18–50 years) in generally good health based on medical evaluation, with body mass index (BMI) between 19 and 30 kg/m2 and weight between 50 and 100 kg. Due to a preclinical signal suggesting the potential for decreases in CD4+ T-cell counts after extended exposure to GSK3732394, participants were excluded if they had CD4+ T-cell count <500 cells/mm3 or CD4+ T-cell percentage outside normal range (32%–64%). Participants were also excluded if they had been exposed to a live vaccine or immune-modulating medication within 1 month of screening or treated with biologic agents within 3 months or five half-lives of dosing.
Procedures
Participants were screened within 30 days before scheduled dosing. After screening, eligible participants were admitted to the clinic on Day - 1. In Part 1, participants were randomized 3:1 to receive a single subcutaneous dose of GSK3732394 or placebo on Day 1. At the start of each single ascending dose cohort, two participants served as sentinel participants and received either blinded GSK3732394 or placebo. Participants remained in the clinic through Day 14 and returned on Days 17, 21, 24 and 28 for follow-up assessment. If terminal half-life was longer than predicted in Part 1, PK/PD and laboratory assessments were planned to occur every 4 days until an estimated five half-lives elapsed.
Pharmacokinetic stopping criteria for dose escalation in Parts 1 and 2 were to be applied if any participant had exposures greater than the no-observed-adverse-effect level (NOAEL) of 93.9 μg/mL for Cmax.
Endpoints and assessments
The primary endpoint was safety assessments in all participants who received ≥1 dose of GSK3732394 or placebo. Safety analysis included adverse events (AEs; assessed continuously, including injection site assessment); vital signs (pre-dose; 1, 2, 4, 8 and 12 h after dosing on Day 1; once daily thereafter); laboratory assessments (Days 2, 3, 5, 8, 11, 14, 17, 21, 24 and 28); electrocardiograms (4 and 12 h after dosing on Day 1; Days 2, 8, 14, 17 and 28, based on expected tmax levels); and physical examinations (Days 2, 8, 14, 17, 21, 24 and 28).
Secondary endpoints were PK assessments, RO profile and immune responses (change from baseline in CD4+ T-cell count and percentage and titres of anti-GSK3732394 antibodies) after single or multiple doses of GSK3732394. To assess GSK3732394 plasma concentrations and CD4 RO, blood samples were collected pre-dose; 0.5, 1, 2, 4, 8 and 12 h after dosing on Day 1; once daily through Day 14; and on Days 17, 21, 24 and 28. Serum levels of GSK3732394 were analysed via electrochemiluminescence immunoassay (GlaxoSmithKline, version 3.1). GSK3732394 was captured with biotinylated PRD828, a helical bundle peptide that binds the anti-gp41 peptide component of GSK3732394 [6]. Captured GSK3732394 was then detected with a Sulfo-Tag CD4 adnectin–specific antibody. Venous blood samples were also collected for CD4 RO. CD4 RO on T cells was analysed via flow cytometry (Q2 Solutions, Morrisville, NC). Fluorescence levels of the CD4-binding adnectin portion of GSK3732394 conjugated to Alexa Fluor 647 (ThermoFisher Scientific, Waltham, MA) were used to calculate the percentage of unbound and bound CD4 receptors. Serum samples for anti-GSK3732394 antibodies were collected on Days 7, 10, 14, 21 and 28; analysed via electrochemiluminescence immunoassay by capturing with biotinylated GSK3732394; and detected with ruthenylated GSK3732394 using a stepwise acid dissociation bridging immunoassay method.
An exploratory analysis included modelling the relationship between GSK3732394 exposure and CD4 RO, the only observable efficacy signal in participants without HIV. The concentration of GSK3732394 that resulted in RO EC50 (equivalent to the concentration required for 50% CD4 RO) was determined.
Statistical analysis
Adverse events were tabulated using Medical Dictionary for Regulatory Activities (version 23) preferred terms; summary statistics were used for laboratory and vital signs results. Severity of AEs, including injection site reactions (ISRs), was determined by the on-site investigator using Division of AIDS AE grading tables (version 2.1; Supplemental Table S1).
Pharmacokinetic/pharmacodynamic analyses were performed using SAS® version 9.4 (SAS Institute, Cary, NC).
A mixed-effects analysis of the emerging PK/PD data was performed as data were available at the end of the cohort. This served to characterize GSK3732394 PK and its link to CD4 RO. This analysis correlated response (RO) with observed GSK3732394 concentrations while adjusting for placebo-associated alterations in CD4 expression levels (e.g. daily rhythms).
A mixed-effects one-compartment PK model with a lag time (Tlag) for absorption from the injection site was fitted to the GSK3732394 concentrations to help define the PK portion of the combined PK/PD model. The PD portion of the PK/PD model required four steps to adjust for the difference from placebo and time effects: (1) mean RO (bound CD4 receptors) among intra-cohort participants who received active drug versus placebo was estimated; (2) using a linear mixed-effects model (adjusting for time of drug diffusion from interstitial space into plasma [Tlag]), mean RO in the active group was modified by amount of placebo-associated alterations in CD4 expression levels; (3) modified mean RO value was back-transformed into a ratio of bound to unbound CD4 among active participants within each cohort; and (4) the PK/PD relationship was derived by correlating bound to unbound CD4 ratios with their corresponding GSK3732394 concentrations using non-linear mixed-effects exposure-response analysis. The model, once validated against observed data, was used to anticipate RO with higher GSK3732394 doses.
Results
Study population
The study was terminated after Cohort 3 of Part 1; therefore, results are limited to the initial three single ascending dose cohorts (10, 20, 80 mg). The doses following the initial starting dose were determined by safety events after administration of the 10-mg dose (for determining the 20-mg dose) and maximum dose elevation allowed (4-fold increase for the 80-mg dose).
Demographics and baseline characteristics of the safety population.
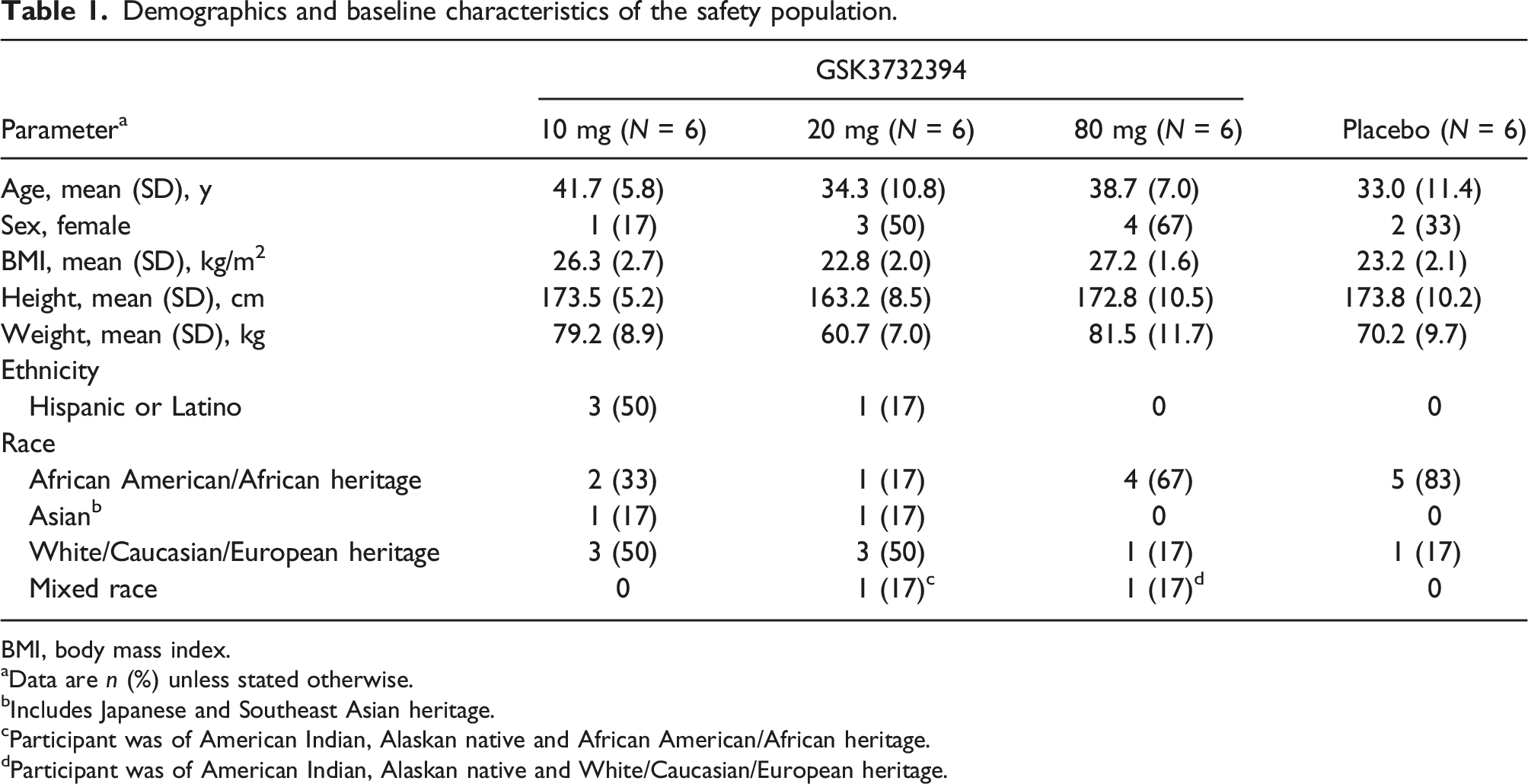
BMI, body mass index.
aData are n (%) unless stated otherwise.
bIncludes Japanese and Southeast Asian heritage.
cParticipant was of American Indian, Alaskan native and African American/African heritage.
dParticipant was of American Indian, Alaskan native and White/Caucasian/European heritage.
Safety
Summary of AEs.

AE, adverse event.
Few moderate (Grade 2) or severe (Grade 3) AEs were observed. One participant who received 10 mg of GSK3732394 had Grade 2 increased transaminases, reported as potentially treatment-related. The participant otherwise remained asymptomatic and the event self-resolved after 14 days. One participant who received 20 mg of GSK3732394 had a Grade 2 generalized drug rash beginning on Day 2. Dermatology consult diagnosed the rash as allergic cutaneous drug eruption. Punch biopsy was performed, and the participant was prescribed 0.1% triamcinolone cream. The rash resolved completely by Day 7. On routine haematoxylin and eosin staining, there was a non-specific, sparse perivascular and interstitial infiltrate of lymphocytes, histiocytes, and occasional eosinophils and neutrophils. One participant in the placebo group had a Grade 3 elevation in serum amylase and was otherwise asymptomatic. No other laboratory abnormalities of at least Grade 2 severity, including clinical chemistries (e.g. changes in absolute or percentage of CD4+ T cells), or significant changes in vital signs were reported. All other AEs observed were Grade 1.
PK/PD analysis
PK
Pharmacokinetics after administration of GSK3732394.

AUC0-∞, area under the concentration-time curve from time 0 (pre-dose) extrapolated to infinity; AUC0-t, area under the plasma concentration-time (t) curve from 0 to t; CL/F, apparent clearance; Cmax, maximum observed concentration; CVb, between-participant coefficient of variation; t1/2, apparent terminal phase half-life; tmax, time of occurrence of Cmax.
aN = 5.
PD: CD4 RO
Unbound CD4 receptor data were measured for the pooled placebo participants and the three treated cohorts (Supplemental Figure S2). These data were then adjusted to account for placebo and time trends, providing an individual data set of CD4 RO percentages per participant per cohort (Figure 2A). The PK/PD modelling performed at the end of each cohort accurately fit the observed PK and CD4 percent RO data and reliably anticipated PK/PD within subsequent dosing cohorts (Figure 2B).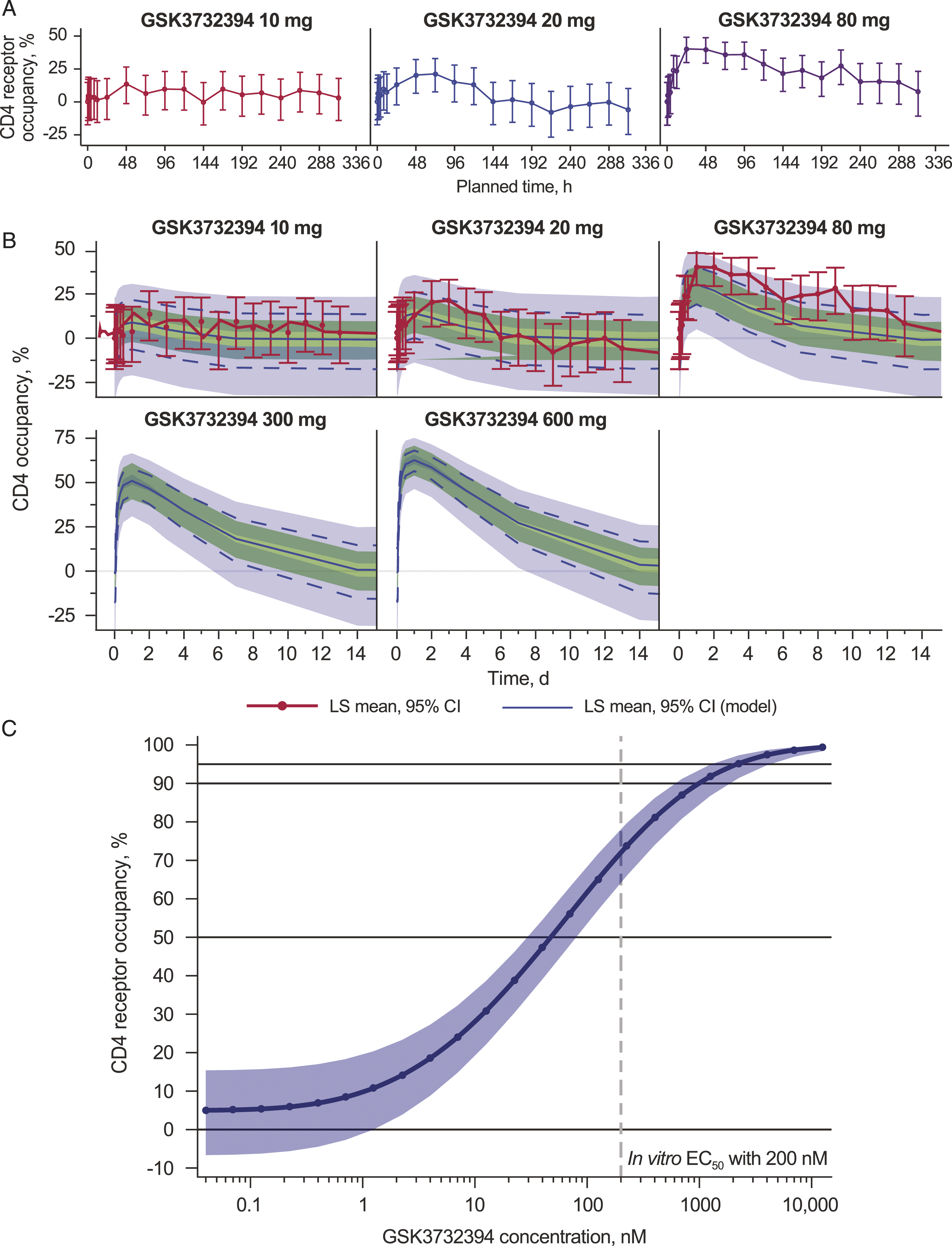
The exposure-response model characterized the GSK3732394 concentration to percent CD4 RO response relationship as RO EC50 (55.6 [SE, 11.2] nM; Figure 2C). This value was comparable to the in vivo mouse model estimate of 15–25 nM and the in vitro estimate of 200 nM. The model and its parameters allowed the assessment of higher doses and the estimation of the feasibility of achieving the desired CD4 RO (≥90%) at trough concentrations. Weekly dosing of GSK3732394 800 mg (maximum allowable dose within the study as determined by active drug concentration [100 mg/mL], greatest number of permissible injections per dosing [4], and highest volume per injection [2 mL] limits) was predicted to not meet the targeted trough occupancy rates. The predicted 600-mg dose would only achieve an average CD4 RO of ∼25% at Day 7 trough. An estimated required weekly dose of ∼2000 mg was predicted as necessary to reach CD4 RO of 90%.
Immunogenicity
Antidrug antibodies were common across all three cohorts (Supplemental Table S3). No pattern between antidrug antibodies and drug exposure or AEs was observed.
Discussion
GSK3732394 was designed as a multi-specific biologic inhibiting virus entry to test the hypothesis of whether a single molecule with three independent inhibitory mechanisms could be used as a complete antiretroviral regimen for treatment or prophylaxis. Linking three inhibitors in the same molecule allowed for multiple synergies that created a highly potent, broad-spectrum agent. Because the major driving force for the synergistic inhibitory potential was the anti-CD4 adnectin binding to its target, CD4 RO was identified as the key parameter for obtaining efficacy, with 90% RO as the target. Preclinical PK evaluation and modelling initially suggested that a weekly subcutaneous 200-mg dose of GSK3732394 would be enough to reach this RO in humans.
This Phase 1, first-time-in-human study in healthy participants evaluated the safety and PK of GSK3732394. Across the three doses tested (10, 20, 80 mg), GSK3732394 was generally well tolerated. No withdrawals due to AEs, serious AEs or deaths occurred. At the completion of each cohort, PK/PD analysis was performed to determine the dose for the subsequent cohort, limited by protocol to a maximum 4-fold increase. Modelling was also used to evaluate the likelihood that the required trough CD4 RO could be obtained with a dose of up to 800 mg of GSK3732394. This maximum dose was chosen based on the maximum volume tolerated for subcutaneous injection (based on GSK3732394 solubility in dosing solution, number of injections and volume per injection). The PK/PD data in humans through Cohort 3 of the single ascending dose study suggested that a weekly dose of 2000 mg would be needed to meet the 90% CD4 RO target. Thus, based upon data at the completion of Cohort 3, the study was terminated, as it was deemed that the investigational compound was unable to achieve its targeted use profile (once weekly, self-administered ART regimen for HIV-1 treatment).
Interestingly, the PK/PD profiles observed were within the expected 2-fold anticipated range of PK/PD performance, supporting the preclinical PK/PD profile. Allometric scaling of monkey and transgenic mouse (expressing human FcRn) data forecasted a human half-life of 50 h (with an expected 2-fold variance). In comparison, the geometric mean half-life for albumin-enhanced therapeutic proteins is 62 h and for recombinant HSA is 19 days [10], suggesting that GSK3732394 had less-than-optimal clearance and exposure compared with most HSA-containing biologics. Preclinical pharmacology yielded potency estimates for CD4 RO in vivo and in vitro. Full suppression of HIV-1 titres in the humanized mouse model was estimated with repeat dosing of GSK3732394 to reach plasma trough levels of 15–25 nM, while 90% CD4 RO could be obtained at a 200-nM concentration. Ninety percent CD4 RO was targeted because of the clinical efficacy observed with ibalizumab (>85%–90% CD4 RO) [11,12]. GSK3732394 may have an advantage over ibalizumab regarding minimum CD4 RO levels needed for maximum efficacy, as GSK3732394 can possibly function in trans, inhibiting virus bound to a different CD4 protein than the one the anti-CD4 adnectin is bound against [6]. Thus, <90% RO levels are required to reach maximum efficacy, as it was shown in vitro that at an EC90 concentration, only ∼1.5% of CD4 on the cell surface is bound [6]. However, since the goal was to use GSK3732394 as a complete long-acting regimen composed only of anti-virus entry inhibitors, the ∼90% CD4 RO level was targeted to ensure efficacy with a high barrier to resistance. These data provided PK and PD expectations for GSK3732394 that were relevant to anticipating treatment success in HIV-1 and, uniquely, could be evaluated in healthy volunteers.
The highest dose of GSK3732394 tested, 80 mg, achieved a trough CD4 RO at 1 week of ∼25%, while the projected trough based upon preclinical modelling was ∼70%. Thus, the actual PK/CD4 RO was considerably lower than projected, and to reach targeted trough values, it was estimated that ten 2-mL injections of GSK3732394 per week would be required. Therefore, the project was terminated after the first three dose cohorts.
Not surprisingly, ISRs with GSK3732394 were the most frequently observed AE; all were generally benign in severity and delayed in onset (Days 10–21). This may be attributed to the nature of injectable biologics. Reported incidence of ISRs with biologic agents has ranged from 0.5% to 40% [13]. Delayed ISRs have been observed with other biologics (e.g. etanercept, infliximab) [14,15] and vaccines (e.g. diphtheria, tetanus, acellular pertussis, COVID-19) [16,17]. Delayed ISRs, which can decrease over time, may be mediated by T lymphocytes, although exact mechanisms are unclear [15]. In this study, one Grade 3 ISR occurred ∼2 weeks after injection and resolved without further action; no discontinuations due to ISRs occurred. Further, no relationship between ISRs and dose or level of antidrug antibodies at Day 14 was observed.
No safety concerns based on clinical chemistries, vital signs or laboratory assessments (including changes in absolute and percentage of CD4+ T-cell count) were reported. One participant had increased transaminases in the GSK3732394 10-mg cohort. Elevated transaminase levels have been previously observed in healthy individuals participating in Phase 1 clinical trials, including those randomized to placebo. This is potentially due to changes in diet and reduced levels of physical activity permitted during prolonged inpatient stays commonly associated with early-stage clinical studies [18].
GSK3732394 did not achieve its desired pharmacology but has provided significant data to guide further development of protein-based HIV therapies and validated the evaluation of relevant PD markers in a healthy volunteer population. The PK and PD of GSK3732394 observed were within 2-fold of the average predictions: to achieve CD4 RO EC50, a concentration of 55.6 nM compared with the in vivo preclinical prediction of 15–25 nM (although in vitro concentration was 200 nM) and an elimination half-life of 22–28 h compared with 50 h. This indicates that the approaches chosen provided acceptable approximations of human data – although reinforcing that they are only approximations. The totality of the available data indicated that the intended trial outcome was possible when considering the projected therapeutic potential of the molecule. These data suggested that the probability of successfully achieving the targeted use profile of GSK3732394 was greater than that of failure. Despite its failings after Cohort 3, outcomes validated the use of the humanized FcRn mouse and monkey models for predicting human PK and the use of the transgenic mouse HIV model for assessing pharmacology, suggesting that only minor refinements may be needed to increase their accuracy in future human predictions. The use of PD evaluation was vindicated by helping decision-making that resulted in the study’s early termination. The PK/PD assessment enabled the trial to respond to ‘real-time’ data rather than requiring the entire data readout. It allowed early focus on multiple aspects of clinical development that inform success in subsequent development phases. Future studies will expand on these findings and aim to improve PK profiles for other long-acting biologics designed as multivalent proteins for the treatment of HIV-1.
Supplemental Material
Supplemental Material - A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection
Supplemental Material for A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection by Mark Krystal, Shiven Chabria, Daren Austin, Allen Wolstenholme, David Wensel, Max Lataillade, Judah Abberbock, Mark Baker, Peter Ackerman in Antiviral Therapy.
Supplemental Material
Supplemental Material - A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection
Supplemental Material for A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection by Mark Krystal, Shiven Chabria, Daren Austin, Allen Wolstenholme, David Wensel, Max Lataillade, Judah Abberbock, Mark Baker, Peter Ackerman in Antiviral Therapy.
Supplemental Material
Supplemental Material - A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection
Supplemental Material for A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection by Mark Krystal, Shiven Chabria, Daren Austin, Allen Wolstenholme, David Wensel, Max Lataillade, Judah Abberbock, Mark Baker, Peter Ackerman in Antiviral Therapy.
Supplemental Material
Supplemental Material - A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection
Supplemental Material for A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection by Mark Krystal, Shiven Chabria, Daren Austin, Allen Wolstenholme, David Wensel, Max Lataillade, Judah Abberbock, Mark Baker, Peter Ackerman in Antiviral Therapy.
Supplemental Material
Supplemental Material - A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection
Supplemental Material for A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection by Mark Krystal, Shiven Chabria, Daren Austin, Allen Wolstenholme, David Wensel, Max Lataillade, Judah Abberbock, Mark Baker, Peter Ackerman in Antiviral Therapy.
Supplemental Material
Supplemental Material - A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection
Supplemental Material for A Phase 1 randomized study of GSK3732394, an investigational long-acting biologic treatment regimen for HIV-1 infection by Mark Krystal, Shiven Chabria, Daren Austin, Allen Wolstenholme, David Wensel, Max Lataillade, Judah Abberbock, Mark Baker, Peter Ackerman in Antiviral Therapy.
Footnotes
Author’s note
Editorial assistance was provided under the direction of the authors by Aarthi Gobinath, PhD, and Jennifer Rossi, MA, ELS, MedThink SciCom, and was funded by ViiV Healthcare.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MK, SC, DW, ML, MB, and PA are employees of ViiV Healthcare and may own stock in GlaxoSmithKline (GSK). DA, AW, and JA are employees of and may own stock in GSK.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the ViiV Healthcare.
Data accessibility statement
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.