
Abstract
Background
JNJ-73763989 comprises two hepatitis B virus (HBV)-specific, liver-targeted N-galactosamine-conjugated short interfering RNA triggers, JNJ-73763976 and JNJ-73763924. JNJ-73763989 pharmacokinetics, safety and tolerability were assessed in two phase 1 studies: Japanese (NCT04002752), and non-Japanese healthy participants and chronic hepatitis B (CHB) patients also receiving the HBV capsid assembly modulator JNJ-56136379 and a nucleos(t)ide analogue (NA) (NCT03365947).
Methods
Healthy participant cohorts were double-blind and randomized to receive a single subcutaneous JNJ-73763989 dose (non-Japanese participants, 35, 100, 200, 300 or 400 mg; Japanese participants, 25, 100 or 200 mg) or placebo. JNJ-73763976 and JNJ-73763924 plasma concentrations were assessed over 48 h. CHB patients received JNJ-73763989 200 mg every 4 weeks plus daily oral JNJ-56136379 250 mg and NA in an open-label fashion. Safety and tolerability were assessed through Day 28 (healthy participants) or Day 112 (patients).
Results
Thirty non-Japanese (n = 4/dose; placebo, n = 10) and 24 Japanese healthy participants (n = 6/dose; placebo, n = 6) were randomized. JNJ-73763976 and JNJ-73763924 exposure generally increased in a dose-proportional manner. Mean plasma half-life was 4–9 h. No differences between pharmacokinetic parameters were apparent between non-Japanese and Japanese healthy participants. In the 12 CHB patients, mean JNJ-73763976, JNJ-73763924 and JNJ-56136379 plasma concentrations 2 h post-dose on Day 29 were 663, 269 and 14,718 ng/mL, respectively. In both studies, all adverse events were mild/moderate.
Conclusion
JNJ-73763976 and JNJ-73763924 had short plasma half-lives and exposure generally increased in a dose-proportional manner; there were no pharmacokinetic differences between Japanese and non-Japanese healthy adults. JNJ-73763989 with or without JNJ-56136379 and NA was generally safe and well tolerated.
Introduction
Approximately 292 million people are chronically infected with hepatitis B virus (HBV) worldwide [1], leading to life-threatening cirrhosis and/or hepatocellular carcinomas in 20–30% of patients with chronic HBV (CHB) [2]. CHB infection is defined as persistent detectability of hepatitis B surface antigen (HBsAg) in serum with or without detectable HBV DNA. CHB is associated with an increased risk of hepatocellular carcinoma, hepatic decompensation, liver transplantation and all-cause mortality compared with non-infected individuals [3]. High HBsAg levels may inhibit neutralizing anti-HBsAg antibodies and contribute to exhaustion of HBV-specific T cells [4]. Functional cure (HBsAg loss sustained for at least 6 months off-treatment, with undetectable HBV DNA) [5] has been shown to improve clinical outcomes in patients with CHB [6,7], and is currently considered an ideal outcome of anti-HBV treatment strategies.
Daily oral nucleos(t)ide analogues (NAs) and weekly pegylated interferon subcutaneous injections are the only drug classes currently approved for use in patients with CHB [8–10]. Treatment with NAs results in HBV DNA suppression and improved clinical outcomes; however, high off-treatment relapse rates necessitate lifelong therapy for most patients [11]. While 48 weeks of pegylated interferon therapy can lead to functional cure in ≤10% of patients, long-term treatment with NA is associated with a substantially lower functional cure rate [12,13].
Novel therapeutic approaches targeting different HBV life cycle stages may provide treatment options of finite duration that can achieve functional cure [14]. One of the strategies that are currently in development is the inhibition of HBV protein production using RNA interference (RNAi) [14–16]. RNAi is an endogenous post-transcriptional gene-silencing mechanism that induces sequence-specific RNA degradation to inhibit gene expression. Integral to this mechanism are short interfering RNAs (siRNAs): duplex RNA molecules approximately 21–25 nucleotides in length that are loaded onto the cytoplasmic RNA-induced silencing complex, resulting in translational repression and degradation of complementary target mRNAs [16–18].
JNJ-73763989 comprises two siRNA triggers, JNJ-73763976 and JNJ-73763924. Each trigger is conjugated to N-acetylgalactosamine (GalNAc) in a triantennary configuration to enable liver-targeted delivery via the asialoglycoprotein receptor expressed on hepatocytes [19]. These triggers target all HBV RNA transcripts derived from covalently closed circular DNA (cccDNA) and host integrated viral DNA, reducing levels of HBV pregenomic RNA (pgRNA) and all HBV proteins, including HBsAg.
Another potential therapeutic target is HBV capsid assembly. JNJ-56136379 is a class-N capsid assembly modulator currently in phase 2 trials in patients with CHB [20]. JNJ-56136379 interferes with capsid assembly kinetics, preventing pgRNA encapsidation and the de novo formation of cccDNA by interfering with capsid disassembly, thereby inhibiting viral replication [21]. JNJ-56136379 resulted in potent dose-dependent reduction of HBV DNA and RNA when given as monotherapy for 4 weeks in treatment-naïve HBV patients [22]. In a phase 2 study of JNJ-56136379 in patients with CHB, treatment with JNJ-56136379 in combination with an NA once daily resulted in greater HBV DNA and RNA reductions from baseline at Week 24 compared to NA treatment alone; there was little effect of JNJ-56136379 and/or NA on HBsAg and HBeAg [20].
The pharmacology and toxicity of JNJ-73763989 up to 300 mg/kg has been explored in preclinical single and multi-dose animal studies in mice, rats, and monkeys (data on file, Janssen). The continuing clinical development of JNJ-73763989 includes two clinical trials: a phase 1/2a first-in-human study comprising healthy participants and patients with CHB from New Zealand, Australia and/or Hong Kong (NCT03365947; henceforth the ‘non-Japanese’ study); and a randomized, double-blind, placebo-controlled, phase 1 study in Japanese healthy participants from the United Kingdom (UK) (NCT04002752; henceforth the ‘Japanese’ study). Here, we present pharmacokinetics, safety and tolerability data for healthy participants from these studies and for a cohort of patients with CHB treated with JNJ-73763989 in combination with JNJ-56136379 and NA in the non-Japanese study.
Methods
Participants
Healthy participants eligible for the non-Japanese study were adults aged 18–55 years with a body mass index (BMI) between 19.0–38.0 kg/m2. Those eligible for the Japanese study were adults aged 20–55 years with a BMI of 18.0–30.0 kg/m2 (body weight ≥45.0 kg) and who were Japanese – i.e. have not resided outside of Japan for >10 years with both parents and all grandparents being Japanese. Participants had to be considered healthy based on medical history, physical examination, vital signs, electrocardiogram, and clinical laboratory tests. Full inclusion and exclusion criteria are provided in Additional file 1.
Patients with CHB eligible for the non-Japanese study were adults aged 18–65 years with a BMI of 19.0–38.0 kg/m2 and who had a diagnosis of hepatitis B e-antigen (HBeAg)-positive or HBeAg-negative CHB infection (of >6 months). Key exclusion criteria included acute hepatitis, seropositive for hepatitis C virus, a history of hepatitis delta virus, and infection with human immunodeficiency virus. Full inclusion and exclusion criteria are provided in Additional file 2.
Ethics
Both studies were conducted in accordance with the Declaration of Helsinki, Good Clinical Practice and applicable regulatory requirements. The study protocols were reviewed by Independent Ethics Committees or Institutional Review Boards and any modifications were approved by the Committees. Written informed consent was obtained from all participants.
Study design
The non-Japanese study was a multicentre study in which healthy participants were recruited at multiple sites in Australia and a single site in New Zealand, and the CHB patient cohort was recruited at a single site in Hong Kong. The healthy participant cohorts were randomised, double-blind and placebo-controlled, and participants received a single escalating dose of JNJ-73763989 and were followed for 28 days. The CHB cohort reported here was an open-label evaluation of three JNJ-73763989 doses administered subcutaneously every 4 weeks plus JNJ-56136379 once daily for 12-week. NA once daily was continued or started at Day 1 of the study and continued through the 12-week JNJ-56136379 treatment period and the 12-week follow-up. The Japanese study was a randomized, double-blind, placebo-controlled, parallel, single-centre study conducted in the UK. Healthy participants received a single JNJ-73763989 dose and were followed for 28 days.
Randomization and study treatment
JNJ-73763989 was supplied as single 200 mg/mL vials containing study drug, prepared by the study clinic pharmacy for both studies. Placebo (0.9% saline) injection volume was matched to the corresponding JNJ-73763989 dose volume.
Non-Japanese study
Healthy participants (n = 30) were randomized 2:1 to receive a single subcutaneous dose of JNJ-73763989 (35 mg, 100 mg, 200 mg, 300 mg or 400 mg) or placebo on Day 1 following a 2-h fast. Six participants were randomized to each dose cohort (JNJ-73763989, n = 4; placebo, n = 2). All administrations were performed in the abdomen. The 400 mg dose was administered as two injections at separate injection sites due to the volume. The 35 mg starting dose is less than 1/500th of the 300 mg/kg, ie the highest dose tested, no observed adverse effect level (NOAEL) in monkeys. The maximum tested dose of 400 mg is 1/45th of the monkey NOAEL. Administration began in two sentinel participants in each dose cohort, one receiving JNJ-73763989 and one receiving placebo. If there were no significant safety concerns on Day 2 based on the Principal Investigator’s assessment, the remaining participants in the dose cohort were treated. Based on observations for all participants in a dose cohort through Day 8, dosing began in the next dose cohort at the Data Safety Committee’s decision.
The CHB patient cohort in the non-Japanese study (n = 12) received JNJ-73763989 200 mg on Days 1, 29, and 57, in combination with a once-daily NA (i.e. entecavir, tenofovir alafenamide, or tenofovir disoproxil fumarate) and JNJ-56136379 250 mg orally once daily up to Day 84, both starting on Day 1. All administrations of JNJ-73763989 were done subcutaneously in the upper arm.
Healthy participants and CHB patients were instructed not to take prescription medications that would interfere with study conduct for 14 days prior to study drug administration and throughout the study.
Japanese study
Healthy Japanese participants (n = 24) were randomly assigned to one of the three panels and within each panel (n = 8) were randomized 3:1 to receive to a single subcutaneous JNJ-73763989 dose or placebo on Day 1, approximately 30 min after a standard breakfast. The doses of JNJ-73763989 for Panels A, B, and C were 25 mg, 100 mg and 200 mg, respectively.
Participants must have discontinued all medication at least 14 days before study drug administration, except for paracetamol or ibuprofen, hormone replacement therapy in postmenopausal women, and hormone-based contraceptives.
Assessments
Pharmacokinetic assessments
In healthy participants from both studies, plasma pharmacokinetic profiles of JNJ-73763976 and JNJ-73763924 were assessed up to 48 h after dosing. Blood samples were collected at pre-dose and 0.25, 0.5, 1, 2, 3, 6, 24, and 48 h post-dose in the non-Japanese study, and at pre-dose and 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 36 and 48 h post-dose in the Japanese study. Urine was collected from 0–6 and 6–24 h in the cohorts receiving 100–400 mg in the non-Japanese study to assess renal excretion. For the CHB patient cohort, blood samples for evaluation of JNJ-73763976, JNJ-73763924, and JNJ-56136379 plasma concentrations were collected 2 h post-dose on Days 1, 29 (±2 days) and 57 (±2 days). Plasma samples were analysed for JNJ-73763976 and JNJ-73763924 using fluorescence hybridization with sequence-specific peptide nucleic acid probes followed by liquid chromatography-fluorescence detection (lower limit of quantification [LLoQ] 1.0 and 2.0 ng/mL, respectively). The principle of this technique is described in Wang et al. [23]. Urine samples were analysed for JNJ-73763976 and JNJ-73763924 using liquid chromatography mass spectrometry (LLoQ 50.0 ng/mL for both). Plasma samples for JNJ-56136379 were analysed using liquid chromatography tandem mass spectrometry (LLoQ 10.0 ng/mL).
Safety assessments
The safety and tolerability of study treatments were monitored up to Day 29 in healthy participants and Day 112 in patients with CHB. AEs and serious AEs (SAEs) were recorded from the point at which a participant provided informed consent. Additionally, all participants underwent physical examinations, vital signs, and echocardiogram assessments, and had samples taken at regular intervals for laboratory tests, including biochemistry, haematology, coagulation, and urinalysis.
Data analysis
As the non-Japanese study was a first-in-human and proof-of-principle study, and the Japanese study was an exploratory study, no formal sample size calculations were conducted. All healthy participants who received JNJ-73763989 and had one or more plasma concentration recording were included in the pharmacokinetic analysis (non-Japanese study, n = 20; Japanese study, n = 18). Plasma pharmacokinetic parameters calculated included the maximum analyte concentration (Cmax), time to Cmax (tmax), area under the plasma concentration-time curve up to the last measurable concentration (AUClast), total drug exposure over time (AUC∞), and terminal elimination half-life (t1/2). Pharmacokinetic parameters were estimated using non-compartmental analysis (Phoenix v8.0, Certara, Princeton, NJ, USA).
Pharmacokinetic analysis in the CHB patient cohort (n = 12) was restricted to determination of plasma concentrations only. Descriptive statistics (mean, standard deviation [SD], median and range) were calculated for continuous variables as appropriate.
All participants enrolled in the studies were included in the safety analyses, with data summarized by treatment group for each study using descriptive statistics (n, percentage).
Results
Baseline characteristics
In the Japanese study, 25 healthy participants were randomized. One participant randomized to receive 200 mg JNJ-73763989 could not be dosed due to a device failure and was withdrawn from the study. All 24 healthy participants (JNJ-73763989, n = 18; placebo, n = 6) who received study treatment completed the study.
Summary of baseline demographics for healthy participants and one CHB patient cohort.

aPatients with CHB received JNJ-73763989 200 mg, JNJ-56136379 250 mg and an NA.
bNot including Japanese healthy participants.
BMI, body mass index; CHB, chronic hepatitis B; NA, nucleos(t)ide analogue.
All 12 patients in the CHB cohort reported herein were Asian, non-Japanese, two-thirds were male (66.7%), and the median age was 44.3 years. The median BMI was 24.6 kg/m2. Two-thirds (66.7%) of the patients were HBeAg-negative and 58.3% were NA-experienced at baseline. Eight and four patients used tenofovir disoproxil fumarate and entecavir, respectively, as their NA (in combination with JNJ-73763989 and JNJ-56136379); no patients were on tenofovir alafenamide. Clinical outcomes of this cohort will be presented in a separate manuscript.
Pharmacokinetics
The pharmacokinetic analysis of JNJ-73763976 and JNJ-73763924 in healthy participants included 19 participants from the non-Japanese study and 18 participants from the Japanese study; plasma concentrations of JNJ-73763976, JNJ-73763924 and JNJ-56136379 were also analysed in 12 CHB patients from the non-Japanese study (who all received JNJ-73763989 200 mg, JNJ-56136379 and an NA). The estimated pharmacokinetic parameters for the non-Japanese study (healthy participants) are based on limited data, both in terms of sample size and the time points of pharmacokinetic assessments.
Healthy participants
In the non-Japanese study, it was suspected that one participant received a 35 mg dose of JNJ-73763989 intravenously instead of subcutaneously; therefore, the pharmacokinetic data from this participant was excluded from subsequent pharmacokinetic analysis. Mean plasma-time concentration profiles for JNJ-73763976 and JNJ-73763924 for healthy participants from the two studies are shown in Figure 1, and plasma pharmacokinetic parameters by dose are shown in Table 2. Across both studies, after a single JNJ-73763989 dose, the median plasma tmax of JNJ-73763976 and JNJ-73763924 in plasma ranged from 3.5–6.1 h and 1.5–6.0 h, respectively, across all doses. This was followed by a relatively short mean t1/2 that ranged from 3.7–9.2 h and 3.7–7.5 h for JNJ-73763976 and JNJ-73763924, respectively (Table 2). There were no major differences in the shape of mean plasma concentration-time curves between JNJ-73763989 doses for either trigger (Figure 1). Cmax and the AUC (including AUClast and AUC∞) for JNJ-73763976 and JNJ-73763924 increased in a dose-proportional manner (Table 2). The maximal urinary excretion rate of JNJ-73763989 was observed at 300 mg, which was 41.7% and 35.4% of the total dose of JNJ-73763976 and JNJ-73763924, respectively (Additional file 3). Plasma-time concentration profiles for A) JNJ-73763976 and B) JNJ-73763924 in non-Japanese and Japanese healthy participants. Solid lines represent healthy participants from the non-Japanese study (n = 4/dose) and dashed lines healthy participants from the Japanese study (n = 6/dose). SD, standard deviation. Summary of JNJ-73763989 pharmacokinetic parameters in healthy participants. aFor the participants in the non-Japanese study that received 35 mg of JNJ-73763989 (n = 4), PK data from one participant was removed from PK analysis due to suspicion of accidental intravenous administration instead of subcutaneous administration (n = 3). bFor the non-Japanese study, the reported value for JNJ-73763976 AUC∞ and t1/2 is based on a single healthy participant for each dose. cFor the non-Japanese study, the reported value for JNJ-73763924 AUC∞ and t1/2 is based on: n = 2 (35 mg), n = 1 (100 mg), n = 3 (200 mg) and n = 3 (300 mg). AUC


Comparison between non-Japanese and Japanese healthy participants
Across all dose cohorts, the median tmax of JNJ-73763976 (6.00 vs 3.5–6.1) and JNJ-73763924 (1.5–6.0 vs 2.5–5.0) in plasma showed no apparent differences between non-Japanese and Japanese healthy participants, respectively (Table 2). Mean Cmax was broadly similar for the 100 mg and 200 mg JNJ-73763989 doses in healthy participants from both studies. The mean plasma half-lives of both triggers were relatively short in both non-Japanese and Japanese healthy participants, ranging from 3.7–7.5 h for JNJ-73763989 doses of 25–200 mg (Table 2). Mean AUClast for JNJ-73763976 and JNJ-73763924 were approximately 2-fold higher following a JNJ-73763989 dose of 100 mg in Japanese versus non-Japanese healthy participants, but were similar between groups at the JNJ-73763989 200 mg dose (Table 2). The limited data between 6 and 48 h for non-Japanese healthy participants preclude a more formal comparison of pharmacokinetic parameters between healthy participants from the two studies.
Patients with CHB
In patients with CHB receiving JNJ-73763989 200 mg, JNJ-56136379 250 mg and an NA, the mean (SD) JNJ-73763976 and JNJ-73763924 plasma concentrations 2 h post-dose on Day 1 were 593 (200) and 217 (72) ng/mL, respectively; 663 (302) and 269 (125) ng/mL, respectively on Day 29; and 806 (348) and 326 (145) ng/mL, respectively on Day 57. The mean (SD) JNJ-56136379 plasma concentration hours post-dose on Day 29 and Day 57 was 14,718 (4117) and 13,873 (4067) ng/mL, respectively.
Safety
Treatment-emergent AEs (TEAEs)
A total of 54 healthy participants (n = 30 and n = 24 in the non-Japanese and Japanese studies, respectively) and 12 patients with CHB (all non-Japanese study) were included in the safety analysis. Overall, JNJ-73763989 was generally well tolerated in both studies, in healthy participants and CHB patients alike. There were no deaths, SAEs or TEAEs leading to discontinuation of the study drug.
Summary of TEAEs in healthy participants.
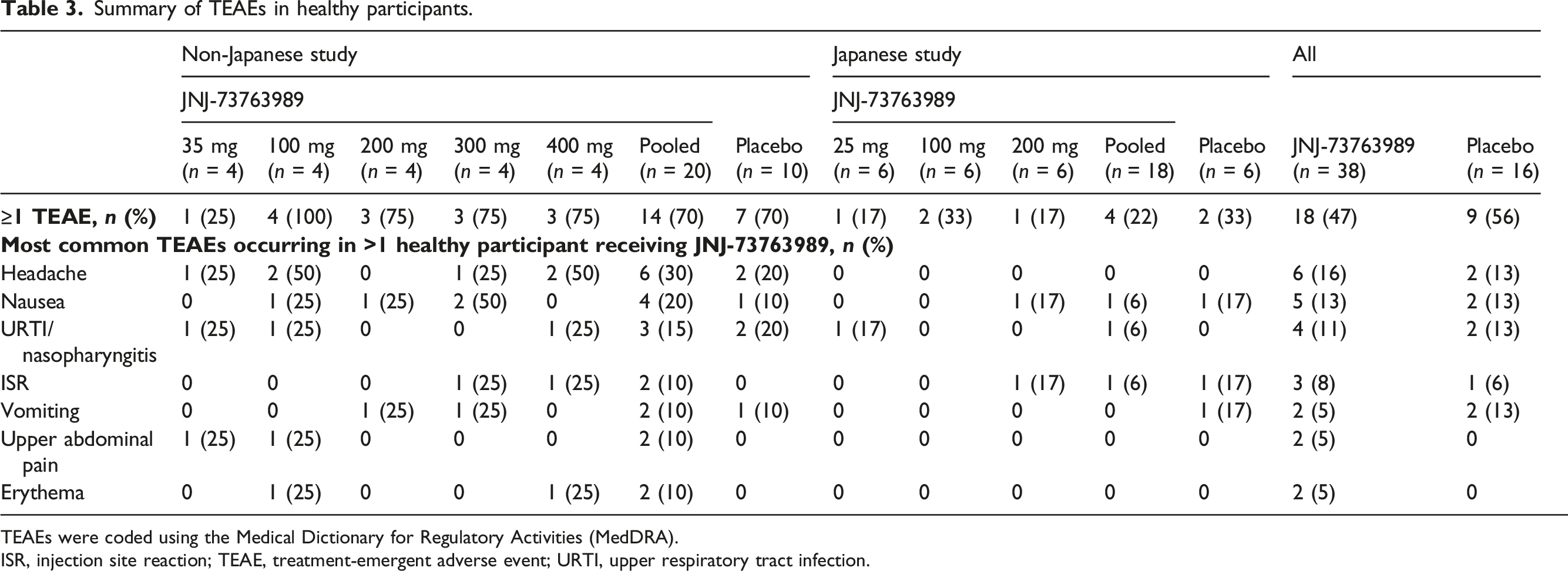
TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA).
ISR, injection site reaction; TEAE, treatment-emergent adverse event; URTI, upper respiratory tract infection.
In the Japanese study, 4/18 (22%) healthy participants receiving JNJ-73763989 and 2/6 (33%) healthy participants receiving placebo experienced at least one TEAE (Table 3). The majority of TEAEs were mild; one healthy participant on JNJ-73763989 (100 mg; musculoskeletal pain) and one receiving placebo (nausea), experienced a Grade 2 TEAE. Treatment-related AEs were reported in 2/18 (11%) healthy participants after receiving JNJ-73763989, which were Grade 1, and 2/6 (33%) healthy participants after receiving placebo, which were all Grade 1 or Grade 2. TEAEs considered treatment related were mainly injection site reaction and nausea. Nausea was reported for two healthy participants (one receiving placebo and one receiving JNJ-73763989 200 mg). All resolved in the next day after injection.
In the pooled population of healthy participants from both studies, 18/38 (47%) healthy participants who received JNJ-73763989 and 9/16 (56%) who received placebo experienced at least one TEAE. The most common TEAEs were headache (16% with JNJ-73763989 vs 13% with placebo), nausea (13% versus 13%) and upper respiratory tract infection/nasopharyngitis (11% versus 13%).
In the CHB patient cohort (12 patients who all received JNJ-73763989 200 mg, JNJ-56136379 and an NA) from the non-Japanese study, 2/12 (16.7%) patients experienced at least one TEAE through Day 112, both were of mild severity and considered not related to treatment. One patient experienced an upper respiratory tract infection and one patient hypertension [24].
Laboratory abnormalities
In the Japanese and non-Japanese studies, no clinically relevant mean changes from baseline over time were observed in any of the haematology, biochemistry, or urinalysis parameters. In the Japanese study, all graded treatment-emergent laboratory abnormalities were reported in at most three healthy participants in any one treatment group. All were Grade 1, except for a Grade 2 glucose increase (JNJ-73763989 25 mg, n = 1/6 [16.7%]), a Grade 2 sodium increase (JNJ-73763989 25 mg, n = 1/6 [16.7%]) and a Grade 2 lipase increase (placebo, n = 1/6 [16.7%]) in a participant who had concurrent Grade 1 amylase increase but no clinical signs or symptoms suggestive of pancreatitis. The most common graded treatment-emergent laboratory abnormalities (occurring in ≥2 healthy participants in any treatment group) occurring in healthy participants receiving JNJ-73763989 and placebo, respectively, were increased glucose (28% versus 50%), decreased phosphate (22% versus 17%), and increased triglycerides (33% versus 17%). Three (50%) healthy participants receiving JNJ-73763989 25 mg had a non-graded direct bilirubin increase.
In the non-Japanese study, there were no clinically relevant laboratory abnormalities in the healthy participants. Grade 1 transient isolated ALT elevations (57–112 U/L) were reported in 5/12 patients with CHB. All elevations were resolved during continued dosing [24].
Discussion
The plasma pharmacokinetics and safety of JNJ-73763989 were assessed in two early phase clinical trials enrolling healthy participants (randomized, double-blind and placebo-controlled) and patients with CHB (with an open-label design) and are reported here. JNJ-73763989 comprises two GalNAc-conjugated siRNA triggers, JNJ-73763976 and JNJ-73763924 that inhibit HBV replication and HBV antigen production through repression of viral RNA translation. Pharmacokinetic analyses following a single JNJ-73763989 dose showed that the mean Cmax and AUC for JNJ-73763976 and JNJ-73763924 increased in a dose-proportional manner. The mean plasma half-lives of both analytes were relatively short (3.7–9.2 h, across all doses), suggesting that JNJ-73763976 and JNJ-73763924 undergo rapid hepatic uptake. This is consistent with the pharmacokinetic profiles of other GalNAc-conjugated siRNAs such as cemdisiran, givosiran and inclisiran which have mean plasma half-lives of 4–10 h [25–27]. The observed median tmax for both JNJ-73763989 analytes, between 1.5–6.1 h, was also consistent with data reported for these siRNAs. Individual renal excretion rates of JNJ-73763989 generally overlapped across all treatment groups, although the mean values increased in a dose-dependent manner, with a mean value of 21–40% excreted in urine.
While limited data for some pharmacokinetic parameters in non-Japanese healthy participants precluded a formal comparison, there were no notable differences in tmax, mean plasma t1/2 and AUClast for JNJ-73763976 and JNJ-73763924 between non-Japanese and Japanese healthy participants. Overall, the data did not reveal any clinically relevant differences in JNJ-73763989 pharmacokinetics between non-Japanese and Japanese healthy participants. Pharmacokinetics of cemdisiran also did not show a difference between Japanese and non-Japanese participants [26]. To our knowledge, there are no reported differences in asialoglycoprotein receptor expression or activity, or enzymes involved in siRNA metabolism (i.e. endo- and exo-nucleases) according to race or ethnicity.
In all participants, there were no deaths, SAEs or TEAEs leading to treatment discontinuation following JNJ-73763989 administration. No dose-limiting toxicities were observed. All TEAEs were mild or moderate in severity and there was no apparent relationship between JNJ-73763989 dose and TEAEs in either study. TEAEs that were at least possibly related to study treatment were reported in 6/20 and 5/10 participants receiving JNJ-73763989 and placebo, respectively, in the non-Japanese study, and 2/18 and 2/6 participants, respectively in the Japanese study. The most common TEAEs were headache (16% with JNJ-73763989 vs 13% with placebo), nausea (13% versus 13%) and upper respiratory tract infection/nasopharyngitis (11% versus 13%). There were no clinically significant laboratory abnormalities reported in non-Japanese healthy participants, and in Japanese healthy participants, laboratory abnormalities were predominantly Grade 1 (other than a Grade 2 glucose increase and Grade 2 sodium increase each in a different healthy participant receiving JNJ-73763989, and a Grade 1 amylase increase in a healthy participant receiving placebo). None of the reported laboratory abnormalities were considered clinically significant. These data show that JNJ-73763989 was safe and generally well tolerated across all doses evaluated.
These early phase clinical studies were proof-of-principle (non-Japanese study) or exploratory (Japanese) in design; therefore, a relatively small number of participants were included. This may affect the generalizability of the findings. While the pharmacokinetic analyses were conducted across six JNJ-73763989 doses in a total of 38 healthy participants, calculation of some pharmacokinetic parameters, for example, t1/2 and AUC
Evaluation of the antiviral activity of JNJ-73763989 is ongoing; significant antiviral activity of the predecessor siRNA, ARC-520, was shown in two clinical trials in patients with CHB [28], as well as in a long-term follow-up study [29]. The phase 2a part of the non-Japanese study described herein is evaluating the antiviral activity of JNJ-73763989, in combination with an NA (with or without JNJ-56136379), in patients with CHB [24]. Separately, large phase 2b studies in patients with CHB, the REEF-1 (NCT03982186) and REEF-2 (NCT04129554) studies, are underway evaluating the safety and efficacy of 48 weeks of combination therapy with JNJ-73763989 and an NA, with and without JNJ-56136379, alongside therapy with JNJ-56136379 and an NA. These studies will provide valuable data regarding the use of combination therapy for the treatment of CHB.
In conclusion, pharmacokinetic evaluation of JNJ-73763989 showed plasma exposure generally increased in a dose-proportional manner and was similar between non-Japanese and Japanese participants. Renal elimination was dose-dependent. A single subcutaneous JNJ-73763989 25–400 mg dose in healthy adults was generally well tolerated. Three JNJ-73763989 200 mg doses in combination with JNJ-56136379 and an NA were also well tolerated in a small cohort of patients with CHB. These results support the further clinical development of JNJ-73763989 for the treatment of patients with CHB.
Supplemental Material
sj-pdf-1-avt-10.1177_13596535221093856 – Supplemental Material for JNJ-73763989 pharmacokinetics and safety: Liver-targeted siRNAs against hepatitis B virus, in Japanese and non-Japanese healthy adults, and combined with JNJ-56136379 and a nucleos(t)ide analogue in patients with chronic hepatitis B
Supplemental Material, sj-pdf-1-avt-10.1177_13596535221093856 for JNJ-73763989 pharmacokinetics and safety: Liver-targeted siRNAs against hepatitis B virus, in Japanese and non-Japanese healthy adults, and combined with JNJ-56136379 and a nucleos(t)ide analogue in patients with chronic hepatitis B by Ed Gane, Man-Fung Yuen, Thomas N Kakuda, Tetsuro Ogawa, Yasushi Takahashi, Nele Goeyvaerts, Isabelle Lonjon-Domanec, Tamisha Vaughan, Thomas Schluep, James Hamilton, Emmanuel Njumbe Ediage, Vera Hillewaert, Jan Snoeys, Oliver Lenz, Willem Talloen and Mich in Antiviral Therapy
Footnotes
Acknowledgements
The authors are grateful to the participants and patients who participated in the studies described. The authors also thank other staff members of Arrowhead and Janssen Pharmaceuticals for their contributions to this study.
Author contributions
All authors were involved in critically reviewing and revising the manuscript for important content and are accountable for all aspects of the work (accuracy and integrity) and approved the final version of the manuscript to be submitted.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Arrowhead Pharmaceuticals. Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Eleanor Coppins, of Ashfield MedComms, an Ashfield Health company, and funded by Janssen Pharmaceuticals.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.