
Abstract
If a planned path reaches a dead-end, one can simply stop. Or one can turn around, walk back to the last intersection and take another path, or one can consider taking few paths in parallel. The last scenario is reflective of the journey of remdesivir, the first antiviral for the treatment of COVID-19, that was approved by FDA less than 10 months after the isolation of SARS-CoV-2, the virus responsible for the COVID-19 pandemic. As of January 2022, 10 million COVID-19 patients have been treated with remdesivir worldwide, but the journey of this molecule started more than a decade earlier with the search for a cure of hepatitis C virus. The development path of remdesivir before the emergence of COVID-19 represents a valuable example of a preemptive pandemic preparedness, but the pursuit of this path would not have been possible without sustaining support of John C. Martin, whom we will sorely miss for his piercing vision, uncompromising leadership, and genuine compassion for patients suffering around the world.
Introduction
John Martin’s medicinal chemistry expertise, his vision as an industry leader, and his determination as a drug developer to bring new medicines to suffering patients was behind the discovery and development of numerous critical antiviral medicines, many of which are nucleoside and nucleotide analogs. Since joining Gilead Sciences in 1990 as the Head of Research and Development until the end of his tenure as the Executive Chairman, John Martin led, supported, and facilitated Gilead’s research, development, and successful approval of the whole portfolio of nucleoside and nucleotide antivirals. These molecules are a rather unique and complex class of pharmaceutical agents because of their challenging synthesis, frequent need for delivery in a prodrug form, multistep intracellular metabolic activation and incorporation into nucleic acids, as well as the potential for adverse off-target effects due to their structural resemblance to natural nucleosides. Because of these challenges, only a few industry leaders pursued these agents seriously as potential medicines. John Martin appreciated not only the challenges, but also the enormous potential of these agents and understood very well how to develop them into meaningful drugs.
As one of the examples from this unique drug class, remdesivir (RDV, Remdesivir and its nucleoside precursor.
Towards hepatitis C virus cure by targeting the RNA-dependent RNA polymerase
Hepatitis C virus (HCV) is associated with a chronic infection of liver that, if left untreated, eventually leads to liver cirrhosis and transplant. The initial standard of care prior to direct-acting antiviral combinations included ribavirin Key nucleoside and nucleotide analogs with antiviral activity against HCV.
In the mid-late 2000s, significant efforts within Gilead were directed towards a search for inhibitors of most of the HCV critical proteins. The approach for targeting RNA-dependent RNA polymerase (RdRp) included both nucleosides and nucleoside phosphonates, but the phosphonates proved very challenging to optimize due to poor viral RNA incorporation efficiency [1]. The 2′-β-C-Me ribose modification was reported to lead to potent purine and pyrimidine nucleoside analogs for HCV [2, 3]. Compound
In parallel, the 1′-CN analog
The ultimate successful approach for improving the selectivity of nucleosides targeting HCV RdRp was the replacement of the 2′-OH with fluorine. Pharmasset scientists synthesized and profiled the 2′-β-C-Me, 2′-F uridine nucleoside, that was initially identified as the metabolite from the corresponding cytidine analog [11]. The active TP was a potent inhibitor of HCV RdRp with a long half-life prompting the exploration of phosphoramidate prodrugs leading to the discovery of
Targeting respiratory syncytial virus and the discovery of remdesivir
RSV, a pathogen unrelated to HCV, impacts multiple groups of patients including pediatrics, elderly, immunocompromised, and those with chronic airway diseases [13, 14]. Treatment options are currently limited to ribavirin
RSV RNA replication and transcription proceeds within a ribonucleoprotein complex containing the RdRp enzyme and inhibition with nucleoside analogs was investigated in parallel to the nucleoside efforts on targeting the HCV RdRp. We conducted a phenotypic RSV antiviral screen of our nucleoside analog library and identified the 1′-CN analog
Broad spectrum in vitro and in vivo antiviral activity profile of remdesivir.
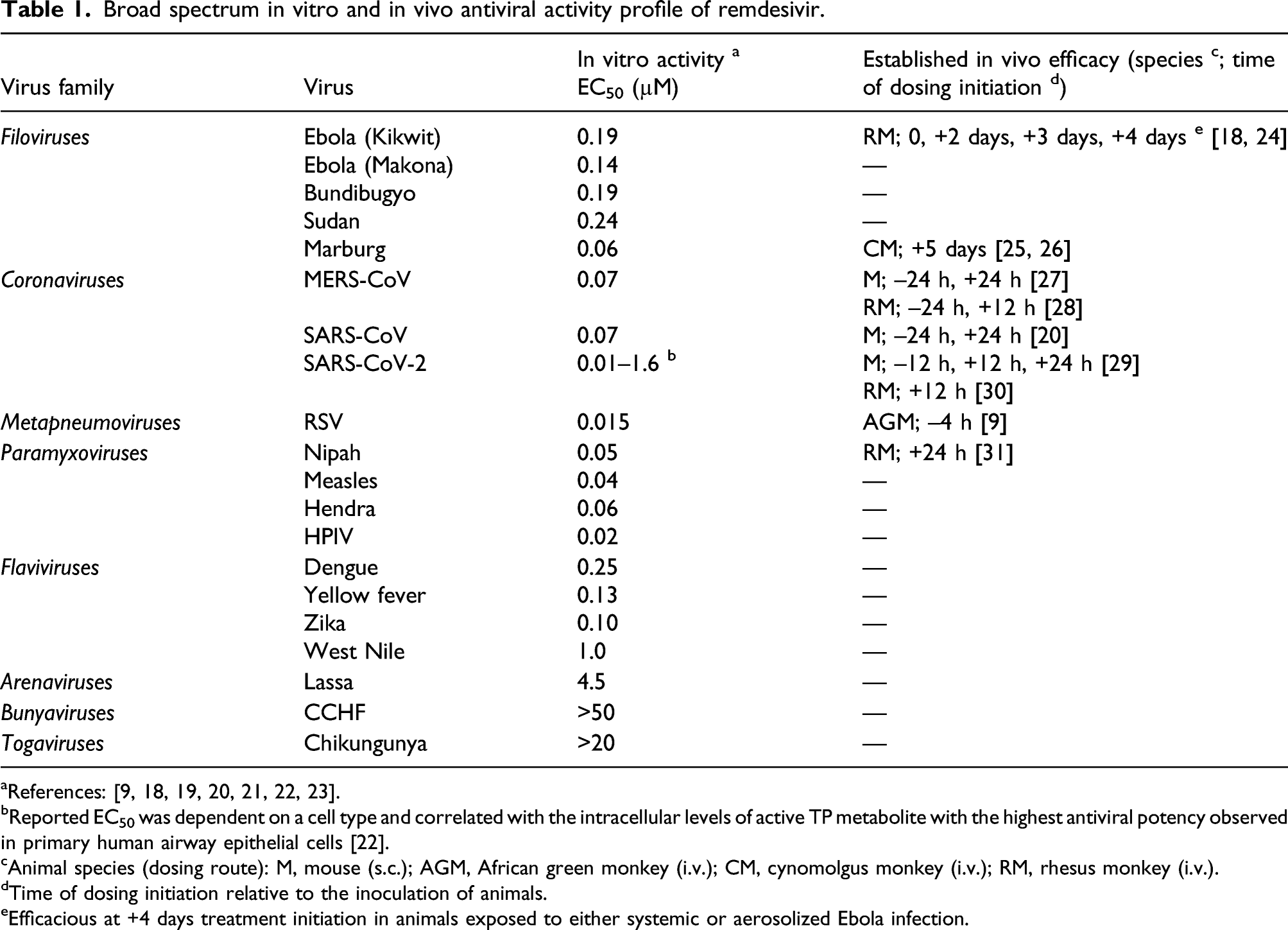
bReported EC50 was dependent on a cell type and correlated with the intracellular levels of active TP metabolite with the highest antiviral potency observed in primary human airway epithelial cells [22].
cAnimal species (dosing route): M, mouse (s.c.); AGM, African green monkey (i.v.); CM, cynomolgus monkey (i.v.); RM, rhesus monkey (i.v.).
dTime of dosing initiation relative to the inoculation of animals.
eEfficacious at +4 days treatment initiation in animals exposed to either systemic or aerosolized Ebola infection.
RDV was evaluated in an RSV infection model in African green monkeys (AGM) to establish in vivo delivery of the active TP metabolite into the appropriate lung cells for driving efficacy. Once daily intravenous (IV) dosing at 10 mg/kg, initiated 4 h prior to infection and continued for 5 days, resulted in a >2.0 log10 reduction in peak viral load in the bronchoalveolar lavage samples relative to vehicle control [9]. A comparatively less robust effect was observed in the nasopharyngeal samples. Pharmacokinetic studies following a 10 mg/kg IV dose in AGMs demonstrated that while the prodrug was cleared rapidly with a systemic half-life of ∼1 h, the active TP in lung tissue samples reached ∼3 μM concentration at 24 h [9]. This concentration of TP was well above the IC50 for RSV RdRp inhibition and supportive of the in vivo efficacy data. Taken together, these results confirmed the ability of RDV to effectively deliver the active TP metabolite with a long intracellular half-life into infected lung tissue, thereby driving efficacy despite a short plasma exposure of intact RDV.
Antiviral activity of remdesivir against other viruses
Ribonucleoside analogs and their prodrugs designed and synthesized at Gilead over several decades, together with analogs acquired through acquisitions and partnerships, formed an ideal library to screen for new antivirals against emerging viruses. Fairly soon after its initial synthesis, RDV was found to exhibit antiviral activity toward Middle East respiratory syndrome coronavirus (MERS-CoV) [32]. While assessing RDV against RSV and other RNA viruses in early 2014, we learned about an unexpected and seriously concerning outbreak of Ebola virus (EBOV) quickly spreading across three neighboring countries in West Africa [33]. Gilead was determined to contribute to the Ebola epidemic response and leveraged its ongoing relationship with the US Centers for Disease Control and Prevention (CDC) by selecting and screening a subset of the assembled nucleoside library for activity against EBOV. This effort resulted in the identification of RDV as the most potent compound. A suite of new phosphoramidate prodrugs was rapidly prepared at Gilead and tested for improved activity through a newly established collaboration with the US Army Medical Research Institute of Infectious Diseases (USAMRIID). However, no new prodrugs significantly differentiated from RDV were discovered for either EBOV [18, 34] or RSV [9].
In early 2015, RDV was selected for the development as potential treatment of EBOV, and in the span of the following 2 years was shown to have a potent broad-spectrum activity toward many other emerging RNA viruses including additional filoviruses (Ebola Sudan, Marburg), paramyxoviruses (Nipah, measles), and flaviviruses (DENV, Zika, YFV) (Table 1) [18, 19]. In particular, further characterization of RDV and its coronavirus (CoV) activity continued with a focus on SARS-CoV, MERS-CoV, and other zoonotic CoVs [19, 20, 21]. The potency of RDV was established in a broad range of human cell types with EC50 < 150 nM for majority of the tested CoVs (Table 1). Minimal to no activity of RDV was detected against arenaviruses, bunyaviruses, and togaviruses [18, 19].
The in vivo efficacy of RDV was also systematically assessed in various animal models of emerging viral infections. Since RDV was found to have substantially limited oral bioavailability, particularly in non-human primates (NHPs) due to high first pass liver clearance, parenteral routes of administration were tested with the main focus on IV administration that resulted in a reproducible and dose-proportional in vivo systemic exposures with desired levels of intracellular TP [9, 18]. RDV has shown consistent in vivo efficacy in various animal models against filoviruses (EBOV, Marburg virus) [18, 24, 25, 26], coronaviruses (SARS-CoV and MERS-CoV) [20, 27, 28], pneumoviruses (RSV) [9], and paramyxoviruses (Nipah) [31], both in the prophylactic and treatment settings (Table 1). In particular, RDV significantly improved the survival in lethal models of filovirus infection in NHPs even when the treatment was initiated 3–5 days post infection (Table 1) [18, 24, 25, 26]. Similar to the in vitro profiling of RDV against emerging viruses, collaborations with the National Institutes of Health (NIH), USAMRIID, and CDC, as well as several academic institutions including the University of North Carolina, Chapel Hill, and the Vanderbilt University were important for conducting a series of in vivo studies from 2015 until the emergence of COVID-19.
The antiviral activity of RDV against multiple RNA viruses is enabled by its unique mechanism of action that results in the selective inhibition of viral RNA synthesis. Inside target cells, RDV is efficiently converted to the TP metabolite via a multi-step metabolic process [9, 18]. The active TP acts as an analog of adenosine triphosphate (ATP) and competes with the natural ATP substrate to inhibit viral RdRps. The primary mechanism of inhibition is the incorporation of the TP into nascent viral RNA by viral RdRps, causing delayed RNA termination [35, 36, 37]. A read-though following the TP incorporation can occur, leading to the presence of the analog in the viral RNA template strand, which very effectively blocks the RNA synthesis via template-dependent inhibition [38]. CoV RdRps were shown to incorporate the TP of RDV more efficiently than natural substrate ATP, and also more efficiently than other viral RdRps such as those from EBOV or RSV [39 and literature references therein].
Remdesivir development from Ebola to Coronaviruses
Shortly after the FDA Investigational New Drug filing in July 2015, a Phase 1 safety and pharmacokinetic assessment of single ascending doses and a repeated dose of RDV administered as an IV infusion was conducted in healthy human adults and demonstrated a consistent dose-dependent pharmacokinetic profile [40]. While the systemic exposures of intact RDV were relatively short (T1/2 ∼ 1 h) due to fast systemic elimination, the high permeability and highly effective metabolic activation in cells and tissues was sufficient to generate high intracellular TP levels in peripheral blood mononuclear cells with a prolonged half-life in excess of 40 h, supporting once-daily administration [41]. RDV has also shown a favorable clinical safety profile with reversible low-grade liver enzyme elevations as the main adverse effect following repeat dosing [40].
Towards the end of West African Ebola outbreak in late 2015, RDV was administered under compassionate use to two patients, one with recurrent Ebola virus disease (EVD) and the other with neonatal EVD. The initial use of RDV occurred in a female Scottish nurse who recovered from EVD, but later experienced reactivation of persistent virus with acute febrile illness, encephalitis, and high levels of EBOV detected in cerebrospinal fluid. Following 14 days of treatment with IV RDV, viral RNA levels became undetectable and the patient recovered [42]. The second compassionate use occurred in a neonate diagnosed with EBOV infection after being born in Guinea to a mother with EVD. The neonate, treated with an investigational monoclonal antibody product and subsequently with IV RDV, recovered from the infection [43]. Before any investigational treatments for EVD were available, EBOV infection was uniformly lethal in neonates [44]. The case in Guinea is the first documented neonate surviving acute EVD.
In 2016, a double-blind, randomized, placebo-controlled, Phase 2 Partnership for Research on Ebola Virus in Liberia (PREVAIL-IV) trial, a U.S.-Liberia joint Clinical Research Partnership, was initiated in male survivors of EVD whose semen continued to be positive for EBOV. The primary objective of the study was to assess clearance of viral RNA from semen to prevent potential sexual transmission of EBOV. In this small trial with a total of 38 men enrolled, 100 mg IV RDV administered once daily for 5 days reduced the presence of EBOV RNA in the semen of EVD survivors 2–6 months after the treatment. The treatment was well-tolerated with no safety issues identified [45].
In 2018, RDV was provided under the Monitored Emergency Use of Unregistered and Investigational Interventions (MEURI) protocol developed by the World Health Organization (WHO) to combat an EBOV outbreak that emerged in 2018 in the Democratic Republic of Congo (DRC) [46]. RDV was subsequently tested in Eastern DRC in an open-label, Phase 2/3 randomized active control trial (PALM Study) that compared three different monoclonal antibody treatments and RDV. The active control group of ZMapp antibody treatment had a 50% mortality rate, which was similar to the RDV group with a 53% mortality rate. The other two antibody products mAb114 and REGN-EB3 demonstrated improved efficacy with mortality rates of 35 and 34%, respectively [47]. The PALM trial was conducted in a region of major political instability and constant security threat that was extremely challenging, with many patients enrolled in advanced stages of EVD, which may explain the reduced efficacy of the experimental therapies compared with preclinical studies. Future EVD therapeutic studies should focus on testing combinations of complementary therapeutics such as RDV and neutralizing antibodies to help identify regimens that could further improve survival in patients with advanced disease and/or aid the clearance of persistent viral reservoirs to prevent potential disease recrudescence. Encouraging efficacy results supporting further exploration of the combination of RDV and neutralizing antibodies have been recently obtained in an NHP model of filovirus infection [48].
In parallel with the compassionate use and subsequent clinical testing in EVD patients, the characterization of RDV activity against other pathogenic emerging viruses continued with an emphasis on the known coronaviruses as one of the most serious potential future epidemic threats due to their highly effective airborne transmission route [49]. Beginning in 2015, a series of preclinical studies were conducted in partnership with experts in the coronavirus field to confirm and further expand the understanding of RDV activity against CoVs (Figure 3). In vitro antiviral testing confirmed potent activity against SARS-CoV and MERS-CoV, and extended these observations to several other zoonotic CoVs with a potential to infect humans. RDV inhibited all tested CoVs and retained antiviral activity across diverse cell types [20, 21, 50]. In addition, both prophylactic and therapeutic efficacy of parenterally administered RDV was demonstrated in rodent and/or NHP models of SARS-CoV and MERS-CoV infection [20, 27, 28]. Based on encouraging preclinical data available by 2017, Gilead decided to explore a possibility of testing the clinical efficacy of RDV in patients with MERS-CoV infection. However, the feasibility of a randomized clinical trial was limited by a low number of MERS cases that occurred almost exclusively in Saudi Arabia, where clinical studies have already been progressing with other investigational agents [51]. This situation dramatically changed during the first days of 2020 with the isolation of SARS-CoV-2 as a novel coronavirus and the causative agent of COVID-19 [52]. Timeline and milestones for the discovery of remdesivir and its preclinical profiling against coronaviruses.
Response to COVID-19 pandemics
Clinical safety data generated during the Ebola treatment program combined with the preclinical data for various CoVs, particularly SARS-CoV and MERS-CoV, supported the early considerations for compassionate use of RDV to treat COVID-19. The first US case of COVID-19 was diagnosed on 20 January 2020. The patient fully recovered following the treatment with supportive care and a course of IV RDV [53]. This first US case opened the international compassionate use program administered by Gilead that utilized the initially available RDV treatment courses [54]. A massive expansion of the drug substance and drug product manufacturing was immediately initiated and supported by a large dedicated team from Gilead and many of its manufacturing partners.
On 6 February 2020, less than a month after the identification of SARS-CoV-2, the first clinical study was initiated in Hubei province in China to test a 10-days course of IV RDV in hospitalized COVID-19 patients [55]. In this randomized, double-blind, placebo-controlled study, the median duration of symptoms before the enrollment was 10 days and the trial did not show overall significant improvement in mortality or time to recovery. However, in patients with symptom duration <10 days, a numerically faster time to clinical improvement was found for RDV treated patients compared to placebo (18 versus 23 days). The trial was prematurely terminated due to a sharp local drop of serious COVID-19 cases, reaching only a low statistical power [55].
A multicenter Adaptive COVID-19 Treatment Trial (ACTT-1) was initiated on 21 February 2020 in the US and several other countries. This randomized, double-blind, placebo-controlled study was sponsored by the National Institute of Allergy and Infectious Diseases (NIAID) in partnership with Gilead and enrolled 1062 adult hospitalized patients with moderate to severe COVID-19 disease [56]. Patients were randomized 1:1 to receive a 10-day treatment of IV RDV or placebo, plus the standard of care. RDV treatment significantly reduced the time to recovery to 10 days from 15 days on placebo (p < 0.001) and was associated with a significant reduction in median hospital length of stay. The mortality rate was significantly lower by day 14 (6.7% versus 11.9%), but not by day 29 (11.4% versus 15.2%) in the RDV treated group. A significant mortality reduction was observed in patients requiring supplemental oxygen at baseline. Serious adverse events were less frequent in the RDV group than in the placebo group (25% versus 32%) [56].
In parallel to the ACTT-1 study Gilead initiated two randomized open-label multi-center randomized SIMPLE studies to compare 5-day and 10-day treatment courses of RDV in hospitalized patients with confirmed SARS-CoV-2 infection. One of the studies (GS-US-540–5773) was conducted in patients with oxygen saturation of ≤94% on room air and evidence of pneumonia [57]. The other study (GS-US-540–5774) was conducted in hospitalized patients with radiological evidence of pneumonia without reduced oxygen levels [58]. Together, the two SIMPLE studies demonstrated the clinical utility of a shorter 5-day treatment course of RDV, a critical finding especially during the early months of pandemics when the drug supply was limited.
A large open-label randomized international SOLIDARITY trial sponsored by WHO was initiated in March 2020 to evaluate RDV and other investigational drugs in >11,000 hospitalized patients from 30 countries. Overall, the rates of in-hospital mortality were similar between the RDV arm and the standard of care arm (12.5% versus 12.7%; p = 0.50) [59], and the mortality rates did not differ among patients with different stages of disease at study enrollment. The SOLIDARITY study protocol was also employed in a parallel open-label, randomized DisCoVeRy trial of >850 hospitalized patients conducted at 48 sites across Europe that found no significant difference between the treatment groups in the disease status on day 15 [60]. Interestingly, in a recent outcome from the Canadian Treatment for COVID-19 (CATCO), a randomized controlled sub-study of the SOLIDARITY trial showed a significant reduction in progression to mechanical ventilation in hospitalized patients treated with remdesivir compared with standard of care [61]. The apparent discordant outcome between the SOLIDARITY and DisCoVeRy trials on one side, and several other studies including the double-blind ACTT-1 study on the other side, triggered a debate about the clinical benefits of RDV treatment [62]. Several experts noted the fundamental differences in the design, protocols, and execution of the different studies that likely led to the differential conclusions, emphasizing the value of stringent double-blind, properly randomized, placebo-controlled clinical trials [63, 64, 65].
In parallel with the clinical testing, preclinical characterization of RDV antiviral activity towards SARS-CoV-2 has also been initiated. RDV has been rapidly established in vitro as a potent inhibitor of SARS-CoV-2 replication in multiple relevant cell types and cultures [22, 23, 66]. In addition, antiviral efficacy in several animal models of SARS-CoV-2 infection has been demonstrated [29, 30] (Table 1).
On 1 May 2020, two days after the release of preliminary data from the NIH ACTT-1 study, FDA granted the Emergency Use Authorization for RDV as the first antiviral treatment for hospitalized COVID-19 patients [67]. In July 2020, the European Medical Agency (EMA) granted RDV a Conditional Marketing Approval followed by a full US marketing approval of RDV by FDA in October 2020 under the commercial name Veklury®. RDV became the first and, as of January 2022, the only direct-acting antiviral drug to achieve full regulatory approval for COVID-19 treatment. This critical milestone in the pandemic response was achieved in less than 10 months after the isolation of SARS-CoV-2 Timeline and milestones for the clinical testing and regulatory approval of remdesivir during the first year of COVID-19 pandemics.
To further examine outcomes of RDV treatment in clinical practice, an extensive real-world data analysis was conducted among US patients hospitalized with COVID-19. In one such cohort, patients starting RDV within 2 days of hospitalization (N=28,855) were matched to unique non-RDV patients (N=16,687). Overall, RDV was associated with a significant reduction in mortality at 14 and 28 days with lower mortality rate benefits observed across all stages of COVID-19 disease [68]. These results complemented findings from ACTT-1 and further supported RDV as a foundational treatment for hospitalized patients with COVID-19. A similar outcome was seen from analyses of two additional independent real-world datasets [69, 70]. In total, the real-world evidence demonstrating the clinical benefit of RDV was collected from nearly 100,000 hospitalized COVID-19 patients [71].
Expanding remdesivir use during pandemics
In order to make RDV available to all patients in need, multiple studies in special populations have been initiated and include testing in pediatric patients, pregnant women, and patients with acute kidney disease. CARAVAN (NCT04431453) is an ongoing open-label study of RDV in hospitalized patients <18 years with PCR-confirmed COVID-19.
Progression of COVID-19 to more severe stages is initially driven by active replication and spread of the virus, which can in turn trigger profound inflammatory process in lungs [72]. To suppress both the viral replication and inflammation, RDV has been tested in hospitalized patients in combination with multiple anti-inflammatory agents. Baricitinib, a JAK1/2 pathway inhibitor, was tested in a double-blind, randomized, controlled ACTT-2 study. When combined with RDV, baricitinib reduced recovery time and accelerated improvement in clinical status of hospitalized patients, particularly those receiving high-flow oxygen or noninvasive ventilation [73]. Based on these results, FDA granted Emergency Use Authorization for baricitinib in combination with RDV in November 2020 [74] and further revised it in July 2021 [75]. Testing of combinations of RDV with other anti-inflammatory agents such as anti-IL23A risankizumab and anti-GM-CSF lenzilumab is currently in progress in ACTIV-5 study in hospitalized patients (NCT04583956, NCT04583969, NCT04988035). Additionally, the NIH ACTIV-1 trial is testing combinations of RDV with infliximab, abatacept, or cenicriviroc (NCT04593940). The ACTT-3 study, on the other hand, found that the addition of IFN-beta 1a to RDV was not clinically differentiated from RDV alone in hospitalized patients [76].
It is generally believed that the benefit of direct antivirals can be fully realized when the agents are used in early stages of acute viral infections. To that effect, a recent double-blind, placebo-controlled randomized PINETREE study in 562 non-hospitalized, high-risk participants with confirmed COVID-19 demonstrated that a short 3-day treatment with IV RDV significantly reduced COVID-19 hospitalization or all-cause death rate by 87% compared to placebo [77]. The 3-day course of IV RDV was safe and well tolerated, leading to FDA approval for use in high-risk non-hospitalized patients with mild-to-moderate COVID-19 [78]. This significant clinical finding also supports the development of oral agents for effectively delivering the RDV active TP metabolite and leveraging the established antiviral mechanism in earlier stages of COVID-19 disease to prevent hospitalizations, particularly among patients with pre-existing risk factors.
The evolution and spread of new SARS-CoV-2 variants with increased transmissibility and/or pathogenicity fueled the spread and multiple surge waves of COVID-19, particularly during the second year of the pandemic [79]. The new variants of concern contain significant genetic changes in the spike protein that is the target for neutralizing antibodies as well as vaccines. In contrast, the RdRp gene (nsp12) of the newly emerged SARS-CoV-2 variants remains genetically stable with only a few mutations located further away from the enzyme active site. Consistent with this observation, RDV retains its full antiviral activity against all major variants of concern including Delta and Omicron [80, 81, 82].
Concluding remarks
The discovery of RDV as a potent antiviral agent with activity against many viruses was driven by collaborative work of multiple cross-functional teams at Gilead over more than a decade. The selection of RDV for clinical development, and the significant efforts applied to the development of formulation and high-capacity manufacturing processes was critical for its rapid regulatory approval and subsequent ability to treat millions of patients globally during the COVID-19 pandemic. We also need to recognize and acknowledge many important partnerships and collaborations that have helped evaluate RDV and its antiviral profile leading to the generation of important preclinical and clinical data that enabled rapid launching of the COVID-19 clinical program at the very beginning of the devastating pandemics.
Overall, the entire discovery and development process of RDV provided a number of relevant lessons important for future pandemic preparedness. The profile of RDV lends support for systemic exploration of nucleosides and nucleotides as a critical part of future antiviral pandemic preparedness strategies because of their broad-spectrum antiviral potential. In addition, fast initiation of efficacy studies with new antivirals during outbreaks of emerging viruses needs to be enabled by preemptive preclinical and early stage clinical development together with the development of a large-scale synthesis and manufacturing process. And finally, productive collaborations and partnerships across all critical sectors including pharma and biotech industry, academia, government, NGOs, and philanthropic organizations should be established early on and sustained throughout the development process.
The RDV program would not have been initiated and advanced without John Martin and his sustained support of antiviral nucleoside research within Gilead for over two decades. We are grateful for his vision and leadership that propelled forward not only the first COVID-19 antiviral drug, but also many other innovative antiviral products, combinations, and treatment regimens that are changing the lives of millions of people around the world.
Footnotes
Acknowledgments
The authors would like to thank Becky Norquist for excellent help with preparation and submission of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.