
Abstract
A concept of “all or nothing” inspired the innovation of a one-pill-once-daily HIV treatment. Atripla® was the one pill that combined efavirenz, emtricitabine, and tenofovir disoproxil fumarate to become the first daily single tablet regimen that forever simplified HIV treatment to enhance patient compliance and thus, sustained viral suppression. The making of Atripla incorporated dry granulation and bilayer compression technologies to achieve stability and bioequivalence in an optimal pill size. In 2011, there lacked a standard of care for chronic hepatitis C infections that was safe, simple, short, free of interferon and ribavirin, and with high cure rates. A fixed-dose combination of ledipasvir and sofosbuvir was developed and approved in 2014 to be the first complete daily single tablet regimen for CHC genotype 1 infection. A spray-drying process for particle morphology engineering in a polymer matrix was used for improving bioavailability.
Introduction
“All or nothing”—a simple, powerful concept coined by John C. Martin had transcended treatment for human immunodeficiency virus infection/acquired immunodeficiency syndrome (HIV/AIDS). Taking this concept to practice was a fulfilling journey made possible by John’s focus on simplifying treatment for people with HIV. The perspective was to treat more people for better health outcomes, to improve medication compliance, to reduce resistance development by avoiding partial regimens, and to enable treatment as a method of prevention.
The advent of highly active antiretroviral therapy (HAART) in the late 1990s led to dramatic decreases in HIV-related morbidity and mortality [1,2], which brought hope of halting the progression and spread of HIV. However, the effectiveness of HAART was not broadly sustained because of the emergence of virologic failure attributable to suboptimal adherence to medications (Figure 1) [3]. It was soon realized that successful treatment with HAART required near perfect adherence to the prescribed regimen [4,5]. Valerie E. Stone, et al. reported findings that “forgot dose” and “feeling sick/side effects” were the most frequent reasons for missing doses of HAART [6,7]. Missed doses and partial regimens allowed viral mutations to cause virologic breakthrough in patients, and also resulted in failure of the secondary prevention of HIV infection from transmission of drug-resistant virus [8]. HAART regimens were complex. They could consist of up to 30 pills daily of multiple medicines, to be taken in different numbers at different intervals, different times, and with or without food. To reduce HIV-related morbidity and mortality, it was imperative to simplify the treatment and radically reduce the complexity of the medication regimens. The degree of adherence was significantly associated with risk for virologic failure (P < 0.001). Adherence of 95% or greater was associated with the lowest incidence of virologic failure. (Reproduced with permission from [3])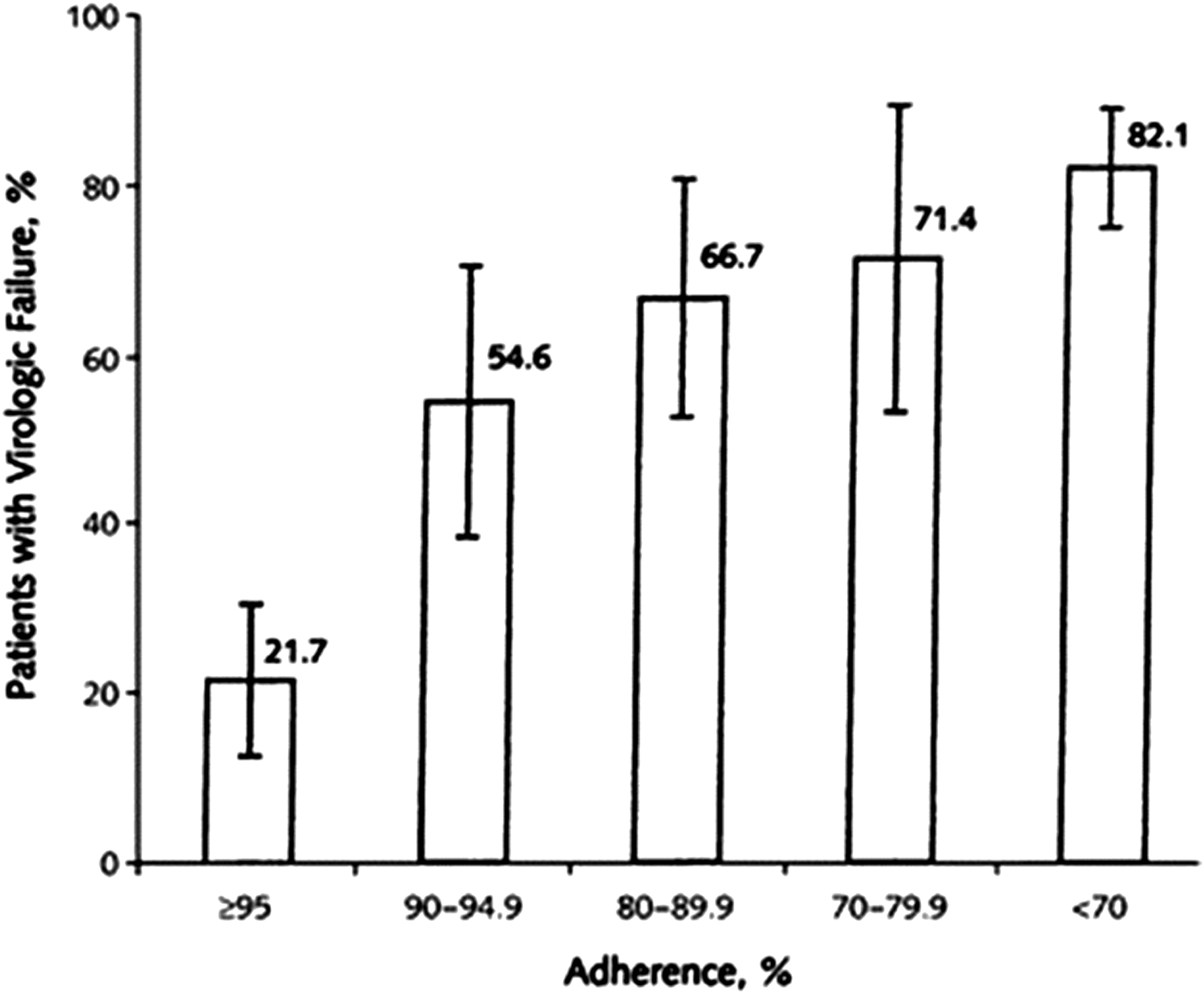
Similarly, successful treatment for hepatitis C infection, which also required multiple-drug regimens, could benefit from treatment simplification to drive medication adherence. The creation of two pioneering single tablet regimens is reviewed herein, one for HIV and the other for HCV infections.
The one pill taken once daily that keeps HIV infection under control
A landmark case of drug innovators and regulatory agencies combatting HIV/AIDS together
Approved in 2006 in the U.S., Atripla® was the first fixed-dose combination that formed a complete regimen from three widely used antiretroviral drugs, to be taken in a single pill once daily to treat HIV/AIDS. 1 This “one-pill-once-a-day” product combined the active ingredients of Sustiva® (efavirenz) a non-nucleoside reverse transcriptase inhibitor (NNRTI) approved in 1998, Viread® (tenofovir disoproxil fumarate or tenofovir DF) a nucleotide reverse transcriptase inhibitor (NtRTI) approved in 2001, and Emtriva® (emtricitabine) a nucleoside reverse transcriptase inhibitor (NRTI) approved in 2003. 2 Emtriva and Viread are also available in a fixed-dose combination known as Truvada® approved in 2004. 3
An unprecedented inter-company cooperation marked the inception of Atripla. This cooperation was between Gilead Sciences, the manufacturer of Emtriva and Viread, and Bristol-Myers Squibb (BMS), the manufacturer of Sustiva, as well as Merck which controlled the marketing of efavirenz in most of the countries outside the U.S. under the brand name Stocrin®. 4 The U.S. Secretary of Health and Human Services, Tommy Thompson, encouraged the industry cooperation while the U.S. Food and Drug Administration (FDA) worked with Gilead and BMS on the clinical pathway for the development of Atripla. Jeffrey Murray and Kimberly Struble of Division of Antiviral Products of the FDA provided regulatory guidance, and Atripla was approved under FDA’s fast track program in 10 weeks following completion of the New Drug Application submission. The NDA was based on bioequivalence with the established individual drugs. Stated in an FDA announcement, the approval of Atripla did not only make the new fixed-dose combination available in the U.S. but also permitted its purchase under the President’s Emergency Plan for AIDS Relief (PEPFAR) program [9]. Andrew C. von Eschenbach, the acting FDA commissioner, said at the FDA press conference, “This offers a particularly important advantage for patients in many countries that are most affected by the AIDS epidemic and will also have a major impact in the U.S.” [10].
Formulation and process design principles
Creating Atripla from three separate drugs carried uncertainties because of the requirements associated with multiple factors: a pill size that would be acceptable to patients, the compatibility of three different active ingredients, the bioequivalence of the combined product to each of the individual drugs, the manufacturability for large-scale production, and the urgency of developing an improved regimen for patients during an HIV epidemic.
The formulation and process design for the combined product required a formulation with an overall high drug load, minimal physical and chemical interactions among the three active ingredients, and adequate stability to support distribution in global climatic zones. It should also be capable of achieving bioequivalence, and must possess good physical and mechanical characteristics for a robust manufacturing process.
Setting a high bar—a complete regimen in a single pill taken daily
To achieve sustained effectiveness of HIV treatment by avoiding partial regimens, the focus was to simplify treatment to one pill of a size amenable to be taken daily as a complete regimen from an effective combination of drugs used in HAART. In other words, developing a complete regimen in a single pill represented the greatest promise for improving the lives of HIV-infected people.
The formulation evaluation began by exploring ways to increase overall drug load to minimize tablet size. A Sustiva tablet containing 600 mg efavirenz weighed 1200 mg, and a Truvada tablet containing 200 mg emtricitabine and 300 mg tenofovir DF weighed 1000 mg. Simply combining Sustiva and Truvada would form a mass of 2200 mg—much too big to be a single pill. Therefore, the formulation had to be modified significantly to reduce the pill size. Gilead set an aspirational goal of a maximum tablet weight of 1600 mg for a single tablet containing three active ingredients that totaled 1100 mg. To achieve this goal, the drug load had to be increased to nearly 70% or higher—a level considerably greater than the drug loads in Sustiva (50%), Truvada (50%), Emtriva (50%), and Viread (45%). Incremental formulation adjustments could not drive such a substantial increase in drug load and it would require fundamental formulation design changes. Yet, a high drug load left little room for formulation design using excipients and process operations to ensure the desirable stability, flow properties, or compressibility of the formulation. More critically, fundamental changes in formulation design would put significant risks on achieving equivalent pharmacokinetics (PK) between the combined product and the individual products.
Biopharmaceutics considerations
Aqueous solubility and intestinal permeability of a drug substance are critical to its biopharmaceutics characteristics [11]. Emtricitabine is freely soluble in water, efavirenz is practically insoluble in water, and tenofovir DF is in between being slightly soluble. The disparity in the intrinsic solubilities spanned a range of four orders of magnitude (from 0.009 mg/mL for efavirenz to 8.5 mg/mL for tenofovir DF to 119 mg/mL for emtricitabine). Considering that one gram of emtricitabine could dissolve in less than two teaspoons of water and one gram of efavirenz would take nearly 30 gallons of water to dissolve, different formulation designs would be required to accommodate the disparate solubilities of the three molecules.
Efavirenz is a high permeability compound with a very low aqueous solubility and, therefore, its PK performance would be significantly limited by the dissolution of efavirenz from the tablet. The formulation design for Sustiva tablets involved formulating efavirenz at a 50% drug load employing high-shear wet granulation with an aqueous solution containing a surfactant to facilitate drug dissolution. In addition, efavirenz was formulated as small particles generated through micronization to increase particle surface area and enhance dissolution. In contrast, because of their high solubilities, emtricitabine and tenofovir DF were not anticipated to pose PK challenges. Truvada was formulated with standard high-shear aqueous granulation of a blended mixture of emtricitabine, tenofovir DF, and excipients. However, a concern for formulating the three drugs together was whether the presence of emtricitabine and tenofovir DF in the microenvironment surrounding efavirenz would affect the dissolution and release of efavirenz from the co-formulated tablet. This uncertainty led to the exploration of bilayer tablets to segregate the drugs in two separate layers. The production drawbacks were that the formulation and manufacturing process for bilayer tablets would be more complex and generally lower yielding than a typical single layer process.
Physical compatibility and chemical stability
In experimenting with a higher drug load for emtricitabine and tenofovir DF using the same wet granulation process as for Truvada, the granulated mixture unexpectedly melted during fluid-bed drying at a temperature around 60°C, which was much lower than the respective melting points of emtricitabine and tenofovir DF. This melting phenomenon had never been observed during production of Truvada, which had a 50% drug load. The melting suggested the formation of a eutectic mixture of emtricitabine and tenofovir DF at a high drug load in the wet granulation. As such, an alternative granulation process was needed and a dry granulation process employing a roller compaction technology was developed to achieve a high drug load for emtricitabine and tenofovir DF.
The main degradation pathway for emtricitabine is hydrolytic deamination of the fluorocytosine moiety; for tenofovir DF, the main degradation pathway is the hydrolysis of the phosphonyl prodrug moiety. A dry granulation is therefore well-suited to ensure good chemical stability of both emtricitabine and tenofovir DF. Another concern to address was the stability impact on emtricitabine and tenofovir DF when combined with a wet-granulated efavirenz formulation containing sodium lauryl sulfate, an ionic surfactant. Again, it would be informative to evaluate segregation of emtricitabine/tenofovir DF granules from efavirenz granules in separate layers.
From design to commercialization
A comprehensive plan emerged from insights gained about the biopharmaceutical, physical, chemical, and mechanical characteristics of the three active ingredients. The approach to the plan was methodical and aimed at exploring design hypotheses, de-risking uncertainties of stability, compatibility and PK performance, and expediting advancement to a one pill, once-daily combination product. In all, more than 70 prototype formulations were designed and evaluated. Formulations of different drug loads, tablet weights, excipients, and granulation processes were evaluated. These efforts uncovered nonobvious challenges requiring innovative solutions [12]. The selection of candidate formulations was guided by targeted screening studies for measurements relevant to PK performance.
Drug load and excipients
Evaluation of formulation composition focused on minimizing tablet size while achieving adequate drug dissolution and absorption at a high drug load incorporating different levels and types of surfactants (ionic and nonionic) and disintegrants.
Granulation and tablet compression
Three distinct manufacturing process approaches were evaluated, and these involved dry and wet granulations as well as single layer and bilayer tablet compression. (i) The first was a co-granulation of efavirenz, emtricitabine, and tenofovir DF by roller compaction dry granulation (Figure 2), and compression of the granules into single layer tablets. This allowed for the smallest tablet size and high manufacturing throughput with an excellent yield. In this dry-granulated formulation of a single layer tablet, good chemical stability was achieved for all three drugs. (ii) The second approach consisted of a bi-granulation process in which efavirenz was wet-granulated with a surfactant, whereas emtricitabine and tenofovir DF were dry co-granulated. The two types of granules were then mixed and compressed into single-layer tablets. It was discovered that mixing the dry-granulated emtricitabine and tenofovir DF with wet-granulated efavirenz containing a surfactant negatively impacted the stability of emtricitabine and tenofovir DF. (iii) The third approach, also a bi-granulation process, differed from the second approach in that the two granulations were kept separate and compressed into bilayer tablets. Good stability was observed for all three drugs. Fundamental concept of a roller compactor. Powder materials are compacted between rollers into ribbon-shaped mass, which is then milled to form dry granules

Screening studies for tablet performance
In addition to chemical stability and physical compatibility evaluations, several key in vitro and in vivo measurements were used to guide the selection of candidate formulations for human PK bioequivalence studies. Focused beam reflectance measurement of the size and size distribution of efavirenz particles in the granulated formulation was found to be a good indicator for the trend of efavirenz dissolution rate. Moreover, in vitro dissolution profiles of efavirenz, emtricitabine, and tenofovir DF in the combined tablets were characterized in a flow-through-cell apparatus using biorelevant dissolution media such as simulated intestinal fluid containing bile salts and phospholipids. This study helped differentiate combination formulations projected to have drug release comparable to the individual reference products. Those prototype formulations exhibiting favorable in vitro results were further studied for PK performance in dogs.
Human bioequivalence studies
Tablet weight, drug load, and bioequivalence outcome of combination products of efavirenz, emtricitabine, and tenofovir DF compressed in (A) single-layer tablet configuration and (B) bilayer tablet configuration.

aFormulation 5 was finalized to be the commercial product, Atripla®.
Racing against time
The development of a combination product of efavirenz, emtricitabine, and tenofovir DF began in March 2004. By December 2004, BMS and Gilead established a U.S. joint venture, the first of its kind in the field of HIV therapy, to develop and commercialize a fixed-dose combination of the three HIV medicines. On this occasion, DHHS Secretary Thompson said, “I am pleased to see the collaboration and efforts of Bristol-Myers Squibb and Gilead. This partnership to create a fixed-dose combination of three HIV medications represents an important advance in our collective effort to deliver simplified therapy for people living with HIV.”
Nineteen months after the joint venture was established, Atripla became the first FDA-approved complete treatment regimen for HIV available in a fixed-dose combination taken once daily (Figure 3). This milestone reflected Gilead’s relentless efforts of designing more than 70 prototype formulations and progressing five formulations to clinical bioequivalence studies. These therapies forever changed HIV infection from being a terminal condition into a manageable chronic health condition and also solidified a strategy of using a highly effective treatment as prevention of transmission. Atripla paved the way for Gilead’s subsequent introduction of five additional single tablet regimens. The latest one-pill-once-daily combination regimen from Gilead was Biktarvy®,
5
which was approved in the U.S. in 2018. Eliminating heavy daily pill burdens of HAART in 1996 with one Atripla® (efavirenz, emtricitabine, and tenofovir disoproxil fumarate) tablet in 2006. HAART: highly active antiretroviral therapy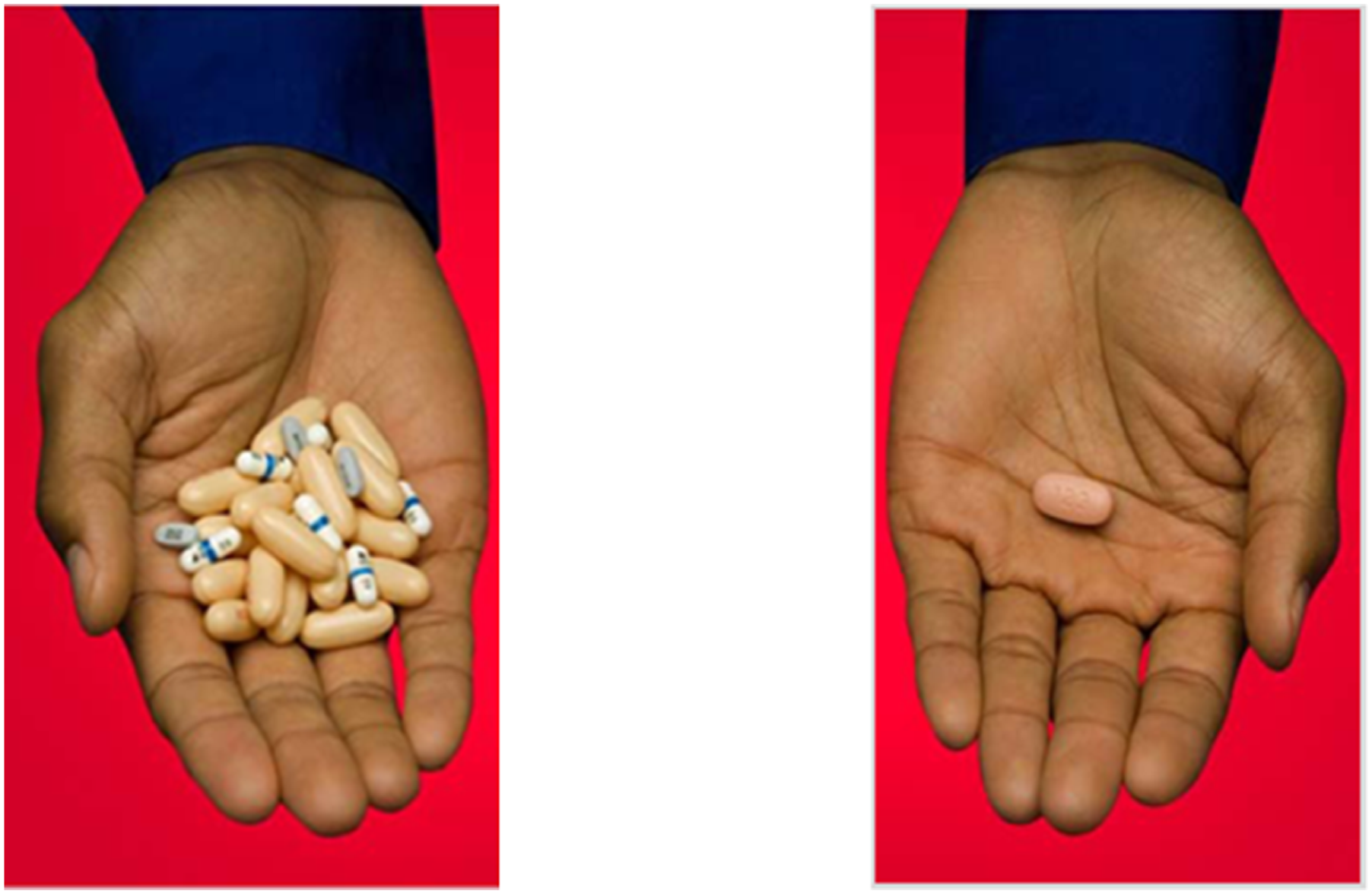
The one pill taken once daily that cures HCV infection in weeks
Up until 2011, the standard of care for treating chronic hepatitis C (CHC) infection for a decade had been pegylated interferon-alpha-2A (peginterferon alpha) weekly injections along with twice daily oral ribavirin tablets for 24 or 48 weeks depending on hepatitis C virus (HCV) genotypes [13]. Sustained virologic responses (SVR) were achieved at 51% in Genotype 1 patients (48 weeks), 82% in Genotypes 2 and 3 patients (24 weeks), and 82% in Genotype 4 patients (48 weeks).
Incivek® (telaprevir)
6
, an HCV NS3/4A protease inhibitor approved in 2011 in the U.S., was indicated in combination with peginterferon alfa and ribavirin for the treatment of Genotype 1 CHC in adult patients. Telaprevir in a regimen of two 375-mg tablets taken three times daily for 12 weeks as a triple therapy (Figure 4) along with 24 or 48 weeks of the peginterferon alfa and ribavirin regimen showed improved treatment outcomes in a head-to-head comparison to a 48-week peginterferon alfa and ribavirin treatment [14]. There were significantly more patients in the telaprevir treatment arm achieving SVR (79% vs. 46%). Moreover, of those achieving SVR on the triple therapy containing telaprevir, a majority (58%) showed extended rapid virologic response allowing their peginterferon alfa and ribavirin treatment to be shortened from 48 weeks to 24 weeks. Shortening the treatment duration was a great benefit to patients. An illustrative dosing regimen of telaprevir, peginterferon alpha, and ribavirin triple therapy 24-week treatment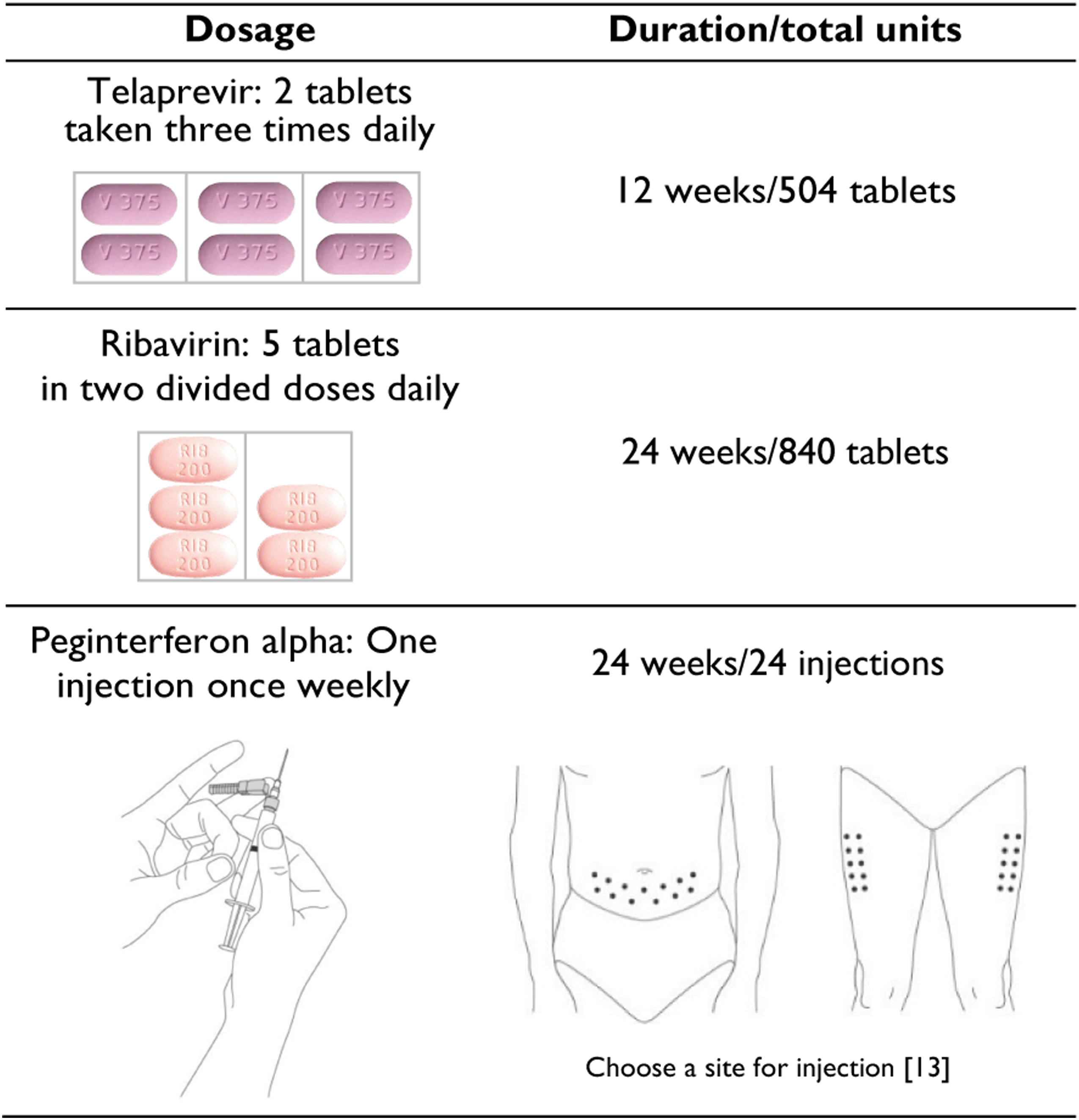
Besides being subjected to a long and burdensome treatment of peginterferon alfa and ribavirin plus telaprevir, HCV patients experienced a high rate of treatment-related adverse reactions including psychiatric reactions, flu-like symptoms, rash, pruritus, nausea, and anemia. Black box warnings highlighted the seriousness of some of the adverse effects [13-15].
Moving beyond interferon and ribavirin
To broadly treat HCV, a complete regimen that was safe, simple, short, free of interferon and ribavirin, and that would deliver high cure rates across multiple genotypes was urgently needed for CHC patients impacted by this debilitating disease. Hepatitis C-associated liver disease and hepatocellular carcinoma were the leading cause of liver transplantation [16]. Many major pharmaceutical companies and biotechnology companies intensified their R&D efforts, racing to deliver the desired HCV cure.
Harvoni—first to arrive
Three years after the introduction of telaprevir as the first direct acting antiviral agent for treating HCV, Gilead’s focus on developing a one-pill-once-daily regimen created Harvoni® as a fixed-dose combination of ledipasvir, an HCV NS5A inhibitor, and sofosbuvir, an HCV nucleotide analog NS5B polymerase inhibitor. 7 Harvoni was the first complete regimen that consisted of a single tablet taken orally once daily for CHC Genotype 1 infection [17].
A molecule that breaks all rules
The “rule-of-five,” derived by C. A. Lipinski, et al. in 1997 [18], predicted poor oral absorption for a molecule with a molecular weight greater than 500, having a partition coefficient greater than 5, and containing more than five hydrogen-bond donors and 10 hydrogen-bond acceptors. Ledipasvir had multiple molecular features and properties defying the “rule” for its high molecular weight (889), high octanol-water partition coefficient (6.9), 8 and high numbers of nitrogen (8) and oxygen (6) in the molecule potentially available for hydrogen-bonding. Therefore, enhancing oral absorption was the first emphasis in developing ledipasvir in combination with sofosbuvir by exploring formulation intervention, crystal form manipulation, and particle engineering.
Crystal form screening
Ledipasvir is a compound with low aqueous solubility and high permeability. It was found to have a low potential to crystallize and was originally isolated as an amorphous free base which showed low oral absorption in Phase 1 clinical studies. Ledipasvir exhibited pH-dependent solubility and was thus susceptible to the influence of gastrointestinal pH and food on dissolution and bioavailability. The development goals were to identify a crystalline form suitable for coformulation with other drugs, capable of mitigating low oral bioavailability and having robust solid-state chemical stability. An extensive crystal form and salt screening study was conducted to evaluate various solvent systems, acidic salts, co-crystals, and solvates. The screening yielded a
Improving bioavailability through spray-drying—a particle morphology engineering
Faced with multiple PK performance challenges for ledipasvir, the formulation scientists launched intensive efforts to explore various formulation approaches starting with lipid-based and surfactant-aided formulations for increasing the dissolution of ledipasvir and its absorption in the gastrointestinal tract with no success. Next, the team examined means of maintaining the poorly soluble drug in a stable amorphous state in the formulation to extend drug supersaturation in solution. This would have the effect of improving the rate and extent of dissolution to enhance bioavailability [19]. Ledipasvir exhibited high solubilities in organic solvents and had good thermal stability which rendered it amenable to the spray-drying process, a technology known to form and stabilize an amorphous form in a polymer matrix for oral and inhaled delivery [20,21]. Engineering designs for particle and crystal form manipulation by spray-drying became integral to the success of drug delivery in the case of ledipasvir.
The crystalline acetone solvate form of ledipasvir was selected to be the drug substance for its consistently good quality and stability in batches from full scale production. In developing the spray-drying process to transform the drug substance into an amorphous dispersion, many approaches were evaluated. Among the options explored were multiple types of polymers including hypromellose, povidone and copovidone, different drug-to-polymer ratios, and different solvent systems. The evaluation was to optimize drug load and particle characteristics such as size distribution, flowability, morphology, glass transition temperature, and process throughput. This comprehensive study identified copovidone to be the preferred polymer for producing an amorphous dispersion of ledipasvir (Figure 5), where acetone along with the process solvent was removed during spray-drying. In a unique finding, there was a single, high glass transition temperature of about 140°C for the ledipasvir–copovidone spray-dried dispersion (SDD), indicating that ledipasvir was well dispersed in the polymer matrix. Therefore, there was an extremely low likelihood for the amorphous ledipasvir to crystalize in the spray-dried dispersion [22]. The presence of crystalline ledipasvir could significantly reduce bioavailability. The absence of phase transition of the amorphous form in the drug product was monitored and demonstrated in stability studies. Chemical structure of ledipasvir acetone solvate, and scanning electron microscopy (SEM) images of crystalline ledipasvir acetone solvate and amorphous ledipasvir dispersed in a polymer matrix with copovidone. SEM of ledipasvir-copovidone spray-dried dispersion showing spherical amorphous particles
The in vitro dissolution measurements showed that drug release from tablets containing ledipasvir SDD greatly exceeded that from tablets containing ledipasvir
Making the molecule and creating a resilient supply chain
The molecular structure of ledipasvir bears several features of synthetic challenges. It contains two fluorines, two bicyclic rings, and six chiral centers in a large molecule of 129 atoms. After thorough process development, a safe, scalable electrophilic fluorination at low temperature was incorporated into the process, and the chiral control employed a combination of chiral salt resolution, asymmetric synthesis, and a chiral pool approach. A convergent synthesis of 23 steps from raw materials that are readily available, through a number of well-controlled production of key raw materials and intermediates to the final ledipasvir acetone solvate drug substance, was developed and implemented.
The manufacture of ledipasvir was complex in chemical transformation, which required long lead times while still conforming to pharmaceutical product regulatory requirements. A diverse supply chain including more than 20 suppliers across North America, Europe, and Asia for materials from key raw materials to the final drug substance was established for the ledipasvir acetone solvate drug substance. A similar strategy was applied in constructing the supply chain for the manufacture of the sofosbuvir drug substance, the ledipasvir SDD, and the Harvoni drug product. Together, a resilient supply chain for Harvoni was built on the foundation of the robust manufacturing processes for both drug substances, the SDD and the tablets. The supply chain was enhanced with redundancy at all supply points that are responsive to demand requirements and reliable in data integrity to assure quality, compliance, and capacity for delivering uninterrupted drug supply to patients globally.
Heroes of Chemistry
The development of Harvoni required intensive efforts from the company’s medicinal chemistry, formulation, and process chemistry teams, challenged not just by one molecule, but by two—ledipasvir and sofosbuvir—and by the process of combining them. This team was recognized by the American Chemical Society with a Heroes of Chemistry Award in 2015 [24]. The development of both compounds and their combination occurred almost simultaneously. The team members 9 were motivated by the possibility of delivering a better standard of care to patients worldwide. The end result was the development of a cure for a chronic, life-threatening disease that can be achieved with just one pill, taken once daily, for duration as short as 12 weeks.
Their innovation enabled the development of Epclusa®10 which is a fixed-dose combination of sofosbuvir and velpatasvir, an HCV NS5A inhibitor. Epclusa was approved in less than 2 years following Harvoni as the first and only pan-genotypic curative treatment for genotypes 1, 2, 3, 4, 5, and 6 CHC patients to be taken orally one tablet once daily with or without food [25].
Footnotes
Acknowledgments
Great contributions to the making of the once daily pill as a complete regimen for HIV and for HCV came from the innovations and the guidance to their colleagues provided by Dimitrios Stefanidis, Rowchanak Pakdaman, and Vahid Zia for the formulation design of Harvoni®, Robert W. Scott for the chemical development of ledipasvir acetone solvate, and Mark M. Menning for the formulation design of Atripla®.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.