
Abstract
John Martin’s untimely death in March 2021 was a huge loss for us personally, Gilead Sciences, the company he built over 30 years and the scientific community concerned with antiviral therapies. We wish to honor John’s legacy by retelling the discovery and history of Tamiflu and his contributions to it. Without his vision, persistence, and keen eye for opportunities, Tamiflu would not exist and Gilead’s path would not have been the same. His strategic thinking around the first oral flu drug is still quite relevant today, when we are still in the SARS-CoV-2 pandemic. John explained it simply in an interview with the Science History Institute in May 2020: “…most of my colleagues, we travel with Tamiflu when we go internationally, because it works for treatment and prevention, and hopefully, there will be a solution like that, eventually, for the Covid virus in addition to vaccines. Most people will get a flu vaccine every year, but there is still disease, we need a pill for treatment and prevention.”
Introduction
Influenza infection, commonly known as “the flu,” has plagued mankind for centuries and is a disease that afflicts almost everybody at some point in their life. The flu occurs with a wide range of severity, including death, and that is despite the general availability of annually updated multivalent vaccines and antiviral drugs, most notably generic neuraminidase inhibitors zanamivir and oseltamivir (brandnames Relenza and Tamiflu, respectively, with FDA approvals in 1999) and the more recent polymerase inhibitor baloxavir marboxil (FDA approval 2019).
As of this writing, we are still deep into the Covid-19 pandemic despite the fact that significant parts of the world’s population have been vaccinated. However, easy to take antiviral drugs, especially those for prophylaxis and early infection, have yet to be approved. While influenza vaccines have been in use for over 70 years, their effectiveness has been thwarted by the unique ability of the virus to genetically drift and shift as well as the wide range in host immune competency, especially in the elderly and immune compromised [1]. A typical flu vaccine consists of multiple, attenuated influenza A and B strains found circulating in the Southern hemisphere which may or may not represent the influenza strains defining the next flu season in the Northern hemisphere. While all viruses gradually undergo mutations to survive in the immune-adaptable host (genetic drift), influenza is unique with its segmented RNA genome (see Figure 1 for virus life cycle) allowing recombination of multiple genomes if a host is harboring multiple influenza strains (genetic shift). Influenza type B is unique to humans, but type A has many more host reservoirs, that is, birds, swine, and more. Herein lies the biggest pandemic potential of influenza as those genetic jumps might not be addressable swiftly enough in routine vaccine production. The biggest influenza epidemic was the Spanish flu from 1918 which took the world by surprise and is estimated to have caused around at least 50M deaths worldwide and 675,000 in the United States alone [2,3]. The US number of Covid-19-related deaths has surpassed this number last September [4].

Replication of influenza A viruses in the lung epithelium
As for the last flu epidemic, the H1N1 swine flu from 2009 luckily remained a “faux-pandemic” with about 60M cases in the United States but only 12,000 deaths [5]. Nevertheless, it became apparent that antiviral drugs effective for contact prophylaxis and early treatment are essential when population wide vaccination fails because of timing or ineffective vaccines. During this time, Tamiflu became a household name and earned record sales of $3.1B in 2009 for its producer Roche, dwarfing typical sales of around $500M in a normal flu season [6].
Much has been written about Tamiflu and the >4800 document hits in a recent Pubmed search supports this assertion. But it is the untimely and tragic passing of John Martin [7], Gilead’s longtime chief executive officer, who inspired us to retell the discovery story of Tamiflu. This project is deeply intertwined with John’s first decade of leading Gilead Sciences toward the successful antiviral company that we know today. The story of the scientific discovery has been well covered in the literature, although John Martin’s name would not be found among the list of authors. Nevertheless, his leadership and mission were instrumental for the discovery of Tamiflu. Here we tell the strategic story highlighting John’s insights and vision.
John joined Gilead Sciences as Vice President of Research & Development in late 1990 leaving behind his 6-year tenure at BMS as the director of antiviral chemistry. John had obtained his Ph.D. in chemistry from the University of Chicago in 1978 after which he did a highly visible and successful postdoc at Syntex. There he discovered ganciclovir in 1980 [8], the first drug against cytomegalovirus. Even back then, it was a rare accomplishment for a young scientist and it naturally launched John’s carrier in antiviral drug discovery. One of the authors (SS) had the privilege to have joined John on the journey from Connecticut-based BMS into the emerging biotech scene of the San Francisco Bay Area. While Gilead Sciences was originally founded as an antisense company, and nucleic acid-based therapeutics still featured heavily in the 1992 IPO, that is, the DNA-based thrombin aptamer [9], John came with an early, passionate vision for creating antiviral drugs, especially for HIV. At that time, HIV was still a death sentence and the suffering was impossible to ignore in San Francisco. At the height of the AIDS epidemic, another major political event started to reshape the world, and that was the fall of the Berlin Wall and the unraveling of the Eastern Block. As an expert nucleotide chemist, John Martin had become aware of a series of acyclic nucleoside prodrugs with potent anti-HIV activity, co-discovered by Tony Holy (IOCB) and Eric deClerq (KU Leuven) some time before the end of the Cold War (chronicled in the 2017 book Cold War Triangle by Loeckx) [10]. John Martin had a chance to evaluate this portfolio of potential antivirals as part of the BMS team in the late eighties and established a collaboration with Holy and deClerq. BMS ultimately let this opportunity pass in 1991. Shortly after arriving at Gilead, John rekindled the negotiations with Holy and deClerq which led to the transformative in-licensing of an estate of nucleoside-phosphonates and their prodrugs in 1991. While this meant a new direction for the small research team at Gilead, the nucleic acid focus of the medicinal chemists was a great setup for doing nucleoside chemistry. John quickly built a team that would do further optimization and even scale-up to the much needed clinical quantities. (A detailed account of these efforts can be found in this issue as well.) Much of the research effort was focused on finding prodrugs for these nucleosides and nucleotides with the goal to achieve oral bioavailability. It is not surprising then that Gilead Science’s first clinical project was the development of intravenous cidofovir. This acyclic phosphonate is a selective chain terminator for viral DNA polymerases and as such not an inhibtor of HIV but rather one for cytomegalovirus. The latter was causing widespread, debilitating retinitis in late stage AIDS patients, often ending in blindness. Cidofovir was approved in 1996 for iv usage, but with the fortunate advance of the first HIV protease inhibitors saquinavir and ritonavir around the same time, the majority of AIDS patients did not have to deteriorate to the stage of CMV retinitis any longer.
While John’s team was successful with its first drug approval, cidofovir was not the transformational drug that a small biotech’s first drug was expected to be.
Chasing innovation by targeting the flu
Despite the enormous utility of a nucleoside and nucleotide platform to potentially target any virus, John Martin had a keen eye for closely aligned opportunities. He challenged the small team of antisense chemists to find the minimal DNA oligomer that could inhibit the influenza endonuclease, yielding interesting dinucleotide inhibitors. When that program stalled, John brought back another influenza target from a scientific meeting that seemed primed for small molecule targeting.
Influenza neuraminidase is an essential protein that resides on the surface of the virus particle and it cleaves the α-ketosidically linked sialic acid (neuraminic acid) from the ends of the chains of hemagglutinin (HA). Without this cleavage event, HA is bound to its receptor such that budding virions cannot escape from the cell surface and dissociate from each other as these emerging virions pick up both proteins from the host cells. Therefore, inhibiting the egress and unclumping of virions had been considered an effective strategy toward flu antivirals for decades.
An early glimpse of the inner workings of this enzyme came from the collaborative effort of the laboratories of G. Laver and P. Colman in Australia that yielded the 1.9 Å crystal structure of neuraminidase of influenza A in 1983 [11,12]. This work revealed a homo-tetramer with its subunits folding into the prototypic “beta-propeller,” with six four-stranded antiparallel beta-sheets arranged like blades of a propeller, shown in an updated version in Figure 2. But it was not until 1991, that the small Australian biotech Biota published a patent with potent neuraminidase inhibitors based on the 2-deoxy-2,3-dehydro-neuraminic acid scaffold (DANA, see Scheme 1). Similar compounds had been described as early as 1974 but had been widely ignored because of their lack of in vivo efficacy in animals [13]. Unlike the highly connected and digital world we live in today, a small company like Gilead Sciences may not have noticed a fresh patent, but the fact that GSK licensed the neuraminidase inhibitors from Biota that same year was hard to miss. A tantalizing, 1992 Biota patent detailed the coordinates of a crystal structure of neuraminidase bound to a potent inhibitor of the DANA series [14]. To visualize how this inhibitor bound to its target, these coordinates had to be turned into something readable for the modeling software of the nineties. While that task for the sole computational scientist at Gilead Sciences back then was tedious, it became frustrating when it became clear that the column with the z-coordinates was erroneous. An algorithm was written to augment the incomplete coordinates toward a useful 3D structure. Help came from a 1992 publication of neuraminidase complexed with N-acetyl-neuraminic acid (NANA, Scheme 1) [15]. At that time, depositions of coordinates at the PDB was entirely voluntary, albeit the call for journals to mandate it had been made earlier [16]. Therefore, structural scientists would rely on the images in papers, especially when they were stereoscopic. Typically, those were generated by line-drawing plotters and it was quite common to hand-color heteroatoms and send color photographs for journal submission. One of the original 1992 figures from the Cusack lab is shown in Figure 3, augmented with the subpocket annotation that became common later (the coordinates were indeed deposited with PDBids 1NSB, 1NSC, and 1NSD and released late 1993) [15]. While those coordinates would have been extremely helpful to the Gilead Sciences team, in this pre-internet era, one had to subscribe to the PDB, which meant that the actual database came on a tape once a year and one would hope that the coordinates from the key papers were indeed in there. Neuraminidase crystal structures. (Left) First depiction of the crystal structure of the apo form of influenza neuraminidase. (Figure 1 from 1983 reference [11]. Conserved residues near the sialic acid binding site. ▲, Glu 119, Asp 151, Asp 198, Glu 227, Asp 243, Glu 276, Glu 277, Asp 330, Glu 425. ▼, Arg 118, Arg 152, Arg 224, His 274, Arg 292, Lys 350. ♦, Tyr 121, Leu 134, and Trp 178). (right) Modern depiction of functional tetramer from a 2019 review. (Figure 1 from 2019 ref [40]: NA exists as a tetramer of four identical monomers. Each monomer consists of four distinct structural domains known as the catalytic head, the stalk, the transmembrane region and the cytoplasmic tail. The head domain structure was generated in Pymol using structural information from Protein Data Bank code 4GZX (A/Tanzania/205/2010 N2 NA). Individual head domain monomers are shown in green, gray, purple, and orange. The NA stalk, transmembrane region, and cytoplasmic tail are yet to be resolved and are depicted here as four alpha helices Compounds discussed in article. Original figure 4 from Burmeister et al. describing the cocrystal structure of NANAD53, N-acetyl-neuraminic acid (diagram showing active site residues and water molecules that interact directly with the bound sialic acid (black). Dotted lines indicate hydrogen bonds. Note that no electron density is seen for the sialic acid OH2 group and the ring conformation of the sialic acid is putative)


While NANA, the inhibitor from the 1992 publication [15], is only a weak inhibitor, its bound structure in conjunction with that derived from the Biota patent suggested where the potency gains from Biota’s inhibitors were coming from. The NANA pyranose ring is found in a high-energy boat conformation while Biota’s 2-deoxy-2,3-dehydro scaffold is unstrained, mimicking the putative flat oxocarbenium transition state of the pyranose. But for both scaffolds all interactions of the ring substituents are very polar. The carboxylate group makes strong contacts with three arginine residues in S1 (R115, R373, and R291). The N-acetyl group exhibits both polar (R149) and nonpolar interactions in S3 (W176, I220, and R222). The glycerol moiety makes two hydrogen bonds with E274 in S5 and hydrophobic contacts to A246 and R222 in S4. The NANA pyranose hydroxyl groups exhibit hydrogen bonds to D148 and E119 which are naturally absent for the Biota scaffold.
The two Biota patents mentioned above revealed that DANA and its derivatives had potent in vivo antiviral activity in a mouse model when given intranasally [14,17]. The most potent DANA derivatives exhibit basic groups instead of the 4-HO, that is, 4-amino-DANA and 4-guanidino-DANA. The latter won FDA approval as zanamavir for inhaled therapy, a whopping 8 years later.
This Biota/GSK program had been discussed at an antiviral conference and John recognized a big opportunity. He did not believe that such small molecules could not be turned into oral drugs. He got his small team to tackle the problem at hand which is low permeability and hence poor oral bioavailability, typical for highly polar molecules. While the neuraminidase inhibitors work in the extracellular space, consistent with the intranasal activity of Biota’s DANA derivatives, an oral drug would be more convenient for patients but for that good permeability is essential. The experience with nucleotide prodrugs gave John’s team the confidence to try a novel approach in the face of the competition with a much larger pharmaceutical company like GSK. Different from working in the nucleotide space, the quest for oral neuraminidase inhibitors, which eventually yielded oseltamivir (Scheme 1), was heavily structure-guided and marks the beginning of Gilead Sciences’ Structural Chemistry group (still led by one of the authors, SS).
By late 1993, John had convinced Chuong Kim, a seasoned medicinal chemistry leader at BMS, to join Gilead. He came with fresh ideas of trying carbocyclic designs. Early data for aminobenzoic acid derivatives suggested that different chemical space could be carved out if one was willing to reoptimize the glycerol moiety which was no longer fitting due to the change in the vector from the aromatic carbon [18]. Choung Kim’s extensive synthetic knowledge allowed the preparation of the isosteric cyclohexene where the 1,2-double bond allows for a better transition state mimetic and the sp3 nature of the 3-carbon offers the correct vector. This new lead scaffold allowed complex stereospecific modification of the 5-C bearing the glycerol chain when shikimic acid (trihydroxy-cyclohex-1-ene 1-carboxylic acid) was found as an auspicious chemistry starting point.
Much of this optimization process has been described [18–22], but a few general observations are worth revisiting. Since the DANA type carbon-linkage was considered synthetically difficult, ether and amine linkages had to be explored. The allylether was the pivotal compound that led to the completely hydrophobic 3-pentylether sidechain replacing the highly polar glycerol moiety of DANA type compounds. This sidechain presented the perfect balance for maintaining potency for both influenza type A and B. The potency gain coming from branched alkyl could not be explained with the DANA-bound crystal structure and structural rearrangments in the S4 and S5 pockets had to be assumed. By late 1995 the team had arrived at the candidate molecule GS-4071 and applied for its first patent [23]. What happened in the S4/S5 pockets became clearer when the Stevens lab at UCB solved a cocrystal structure later (PDBid 2qwk) [20].
For those interested in this detail, the 3-pentyloxy chain of GS-4071 leads to reorganization of the amino acid side chains that increase hydrophobic interactions not seen in the DANA-type structures. E274 is turned over toward R222 to form a tight salt bridge, allowing one branch of the pentyl chain to have close contacts with the π-face of the salt bridge plus I220. The other branch seems to be pushing out the proximal amino acid side chains to pack against A244 and the pi-face of the N292 carboxamide. While this was neither the first nor the last example where polar amino acid side chains create a hydrophobic surface area, even today many modeling software packages have not incorporated these orientation dependent properties adequately (see Figure 4). GS-4071 bound to neuraminidase. Only active site amino acids are shown (stick format colored by element) along with GS-4071 in cyan. Hydrogen bonds are shown as green lines. The surface depicts the very back of the active site as “hydrophilic.” (hydrophilicity coloring from blue to brown). This surface was generated with DiscoveryStudio2021. Note that all software and embedded algorithms using simply the amino acid type for property coloring will miss orientation dependent properties that are often exploited in drug design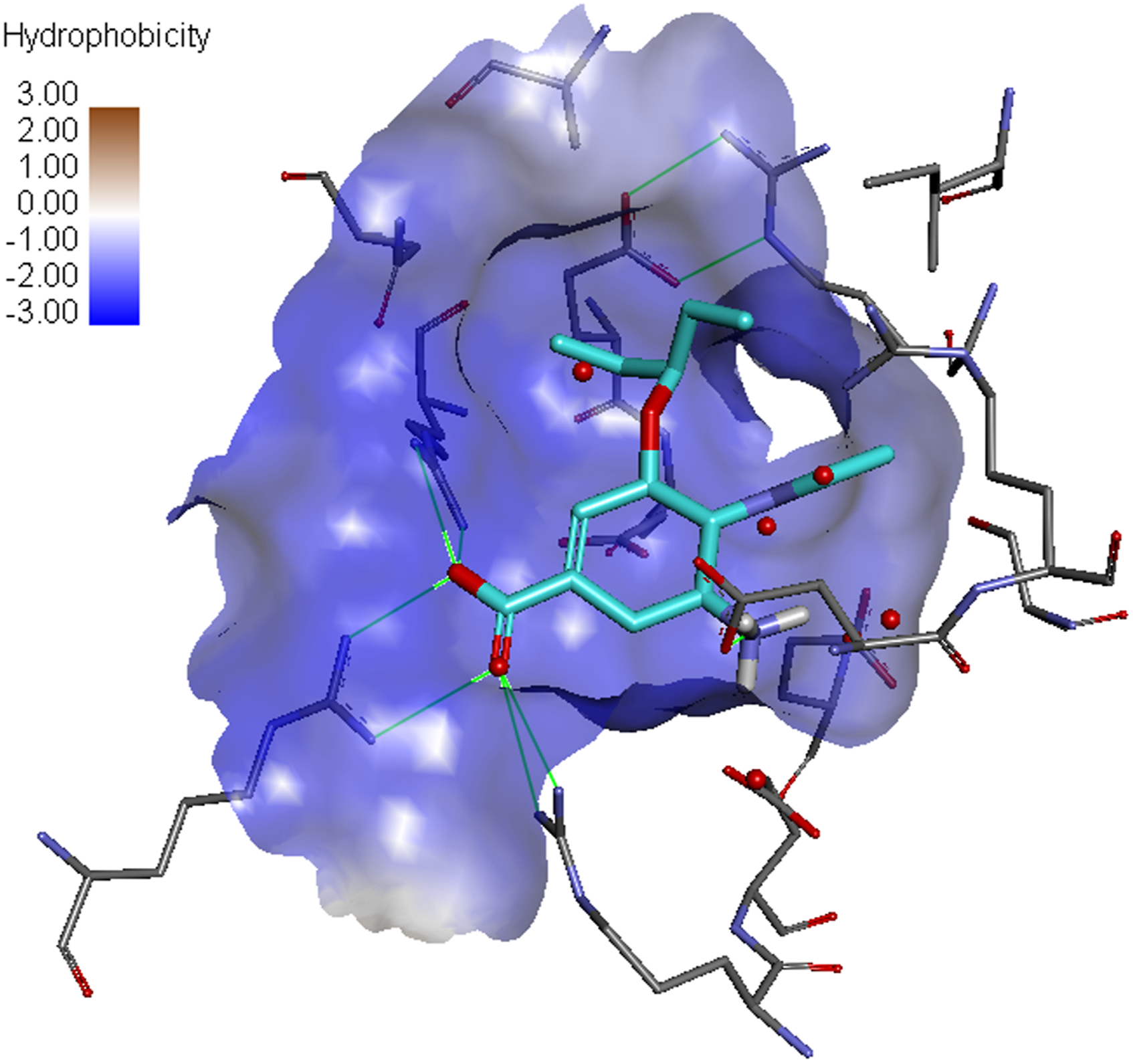
Despite a significant reduction in polar surface area compared to DANA and zanamivir, GS-4071 still exhibited poor bioavailability, but the team knew from the beginning that masking the acid of the zwitterionic molecule would be important. Indeed, oral bioavailability (%F) in rats for the ethylester, oseltamivir, is 35% versus 3% for the parent [24,25]. Note that the even more potent guanidino version of GS-4071 was also tried as an ester, but that %F remained miniscule.
The art of partnering
John knew that Gilead Sciences’ nimble infrastructure, essentially geared toward supporting the HIV antivirals, required a capable partner to develop oseltamivir. Looking far ahead, Gilead would not be readily able to produce the hundreds of kilograms necessary for clinical trials, let alone produce and market a flu drug to patients and their primary care physicians. John’s strategy entailed producing an adequate data package for oseltamivir with minimal internal effort, yet strong enough to convince established pharma companies of its potential for oral efficacy. DMPK studies in mice and rats had been done internally and valuable in vitro antiviral data from the lab of Bob Sidwell at Utah State had generated proof of concept in a mouse influenza challenge study.
In early 1996, potential partners were invited to generate more proof of concept data during their due diligence process. Since the release of a parent acid from an ester prodrug is often highly species dependent, Pfizer, for example, collaborated on further PK studies in ferrets and dogs as well as more challenge studies in mice and ferrets [25]. Later that year, a “worldwide influenza collaboration” with Roche was announced [26]. Here it is worth to note that Franz Humer had just become the new head of Roche’s pharmaceutical division after leaving GSK, where zanamivir was being developed for inhalation. The Roche/Gilead agreement came with $10M upfront payment and an additional $40M for milestones. Roche would fund all research and development at Gilead Sciences and pay significant royalties in the case of approval.
Chasing flu seasons
Strengthening Gilead Sciences’ ability for clinical development, John volunteered Gilead to produce the initial clinical material for the phase 1 study which demonstrated that oseltamivir had promising human PK for twice daily dosing and was well tolerated.
Efficacy studies for acute viral infections are particularly challenging since those drugs should work best when administered shortly after infection or even prophylactically. Therefore, phase 1b studies in humans had to be conducted with experimentally induced influenza infection in healthy volunteers such that treatment can be applied in a time-controlled manner. In such “experimental” influenza phase 1b studies, conducted by Roche in the United Kingdom and Gilead Sciences in the United States, oseltamivir proved to be efficacious in protecting from infection and reducing viral shedding compared to the placebo group in both prophylactic and treatment studies carried out by Hayden and Treanor [27].
Here are some clinical details: oseltamivir (100 mg once or twice daily for 5 days), administered prophylactically, starting 26 h before infection, reduced the number of infections in an 8-day follow-up period from 67% (placebo) to 38% [27] and the number of individuals shedding virus from 50% to 0%. In a treatment study (75 or 150 mg twice daily for 5 days, starting 28 h after inoculation), the quantity of viral shedding (71–96% viral titer reduction compared to placebo) and the time to cessation of shedding were greatly reduced (median time to cessation of viral shedding is 18 h for the oseltamivir versus 96 h for placebo) [28].
While these phase 1b/2 clinical studies with “experimental” flu infection involve “patients” that are indeed infected with influenza, phase three studies must show efficacy, in the “intent to treat” population in the real world. In a typical flu season, even with stringent selection criteria based on the presented symptoms (e.g., fever plus headache, myalgia, and cough), only about two-thirds of the potential patient pool actually have influenza. An additional obstacle for real world efficacy studies comes from the seasonal nature of the infection, hitting each hemisphere only in the cold half of the year. Most efficient flu drug development requires the phase three trials to move around the globe. From 1997 through 1999, a total of four registrational phase three studies were conducted. Two of them assessed oseltamivir efficacy in a treatment setting, the other two in a contact prophylaxis setting. Three of those were run by Gilead Sciences.
Here is more detail about these clinical studies. In the treatment setting, oseltamivir (75 mg twice daily started within 48 h of onset of symptoms) reduced time to alleviation of symptoms by 24 h in the intent to treat population group and by 33 h in the influenza-positive group. The time to resume normal activity was reduced by 2–3 days. In an early versus late start of treatment study (not placebo controlled) with over 1400 patients, early treatment was more advantageous; treatment within 12 h from onset of symptoms reduced the illness duration by 3 days more than treatment initiated 48 h after onset [29,30]. Later studies showed that oseltamivir was efficacious in children and in high-risk patients, that is, the elderly and those with chronic cardiac and respiratory diseases [31]. A few differently designed trials showed efficacy in a prophylaxis setting. In a seasonal prophylaxis study in healthy unvaccinated adults, 75 mg once daily taken for 42 days during a community outbreak reduced the incidence of confirmed influenza from 5% for the placebo group to 1% for the oseltamivir group. Similarly, in a seasonal prophylaxis study in nursing homes with mostly vaccinated seniors, taking 75 mg once daily for 42 days reduced confirmed flu incidence from 4% to <1%. In a study of postexposure prophylaxis in households with an infected adult, 75 mg once daily administered within 2 days of onset of symptoms in the index case and continued for 7 days reduced the incidence of confirmed influenza from 12% in the placebo group to 1% for the oseltamivir group. These studies led to prompt FDA approval of Tamiflu in October 1999 for the “treatment of uncomplicated acute illness due to influenza infections in adults who have been symptomatic for no more than 2 days.” Trial results were published almost concomitantly [27,28,32,33] and reviewed here [34]. A year later, approval was obtained for prophylactic use in adults and a “tutti-frutti flavored oral suspension formulation” for use in the pediatric population [35].
And how did Tamiflu do?
Overall, the “first pill for the flu” was well received and Tamiflu sales quickly exceeded those for inhaled Relenza. Roche’s milestone payments had been instrumental to support Gilead Sciences’ own development pipeline, especially in HIV. But more than just for the financial support Gilead’s contribution in the collaborative development had been well recognized and that softened the blow that came just weeks after the Tamiflu approval. John’s team had to scuttle adefovir for HIV [36] after the advisory board recommended against approval based on safety concerns. Two years later, tenofovir won FDA-approval for treating HIV [37], but without John’s foresight leading to Tamiflu, Gilead’s path would have been very different. Adefovir dipivoxil was eventually approved for HBV at a dose significantly lower than was used in the HIV studies. Years later, John pushed for tenofovir to replace adefovir for HBV, giving patients a better drug despite lower revenues per person treated.
Nevertheless, the story of Tamiflu, which is now generic, would not be complete without mentioning some controversy, common for many widely prescribed drugs. Tamiflu’s efficacy was taunted in the media repeatedly [38] fueled in no small part by the experience of many who accessed the drug (too) late in their infection. Roche’s rebuttal is still worth reading as we are eagerly awaiting the first oral drug(s) for SARS-CoV-2 infection [39]. We hope that the experience with Tamiflu is a lesson for the current pandemic that will lead to treating patients not only as early as possible but also in contact prophylaxis settings.
Footnotes
Acknowledgments
Beyond the few scientists mentioned here directly, there were many more that contributed to the discovery and development of Tamiflu, too many to acknowledge individually. This great Tamiflu community would agree that John Martin played a singular role in bringing this drug to patients.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.