
Abstract
Background
This Phase I, two-part, first-in-human study assessed safety/tolerability and pharmacokinetics/pharmacodynamics of single-ascending doses (SAD) and multiple doses (MD) of the oral toll-like receptor-7 agonist, JNJ-64794964 (JNJ-4964) in healthy adults.
Methods
In the SAD phase, participants received JNJ-4964 0.2 (N = 6), 0.6 (N = 6), 1.25 (N = 8) or 1.8 mg (N = 6) or placebo (N = 2/dose cohort) in a fasted state. Food effect was evaluated for the 1.25 mg cohort following ≥6 weeks washout. In the MD phase, participants received JNJ-4964 1.25 mg (N = 6) or placebo (N = 2) weekly (fasted) for 4 weeks. Participants were followed-up for 4 weeks.
Results
No serious adverse events (AEs) occurred. 10/34 (SAD) and 5/8 (MD) participants reported mild-to-moderate (≤Grade 2), transient, reversible AEs possibly related to JNJ-4964. Five (SAD) participants had fever/flu-like AEs, coinciding with interferon-α serum levels ≥100 pg/mL and lymphopenia (<1 × 109/L), between 24–48 h after dosing and resolving approximately 96 h after dosing. One participant (MD) had an asymptomatic Grade 1 AE of retinal exudates (cotton wool spots) during follow-up, resolving 6 weeks after observation. JNJ-4964 exhibited dose-proportional pharmacokinetics, with rapid absorption (tmax 0.5–0.75 h) and distribution, and a long terminal half-life (150–591 h). Overall, no significant differences in JNJ-4964 pharmacokinetic parameters were observed in the fed versus fasted state. JNJ-4964 dose-dependently and transiently induced cytokines with potential anti-HBV activity, including interferon-α, IP-10, IL-1 RA, and/or MCP-1, and interferon-stimulated genes (ISG15, MX1, and OAS1) in serum.
Conclusions
In healthy adults, JNJ-4964 was generally well-tolerated, exhibited dose-proportional pharmacokinetics and induced cytokines/ISGs, with possible anti-HBV activity.
Introduction
Chronic hepatitis B (CHB) virus infection affects ∼292 million people worldwide [1], with 20–30% of CHB patients eventually developing cirrhosis, liver failure or hepatocellular carcinoma [2]. Approved therapies for hepatitis B, pegylated interferon-α (IFN-α), and nucleos(t)ide analogs (NAs) reduce viremia and improve long-term outcomes [3,4], but rarely lead to functional cure, defined as hepatitis B surface antigen (HBsAg) loss sustained for ≥6 months off-treatment, with or without anti-HBs seroconversion. Hence, life-long therapy with an NA or treatment for 48 weeks with pegylated IFN-α is required for most patients [5–7]. Recognized causes of low cure rates are high HBsAg load, failure of the immune system to recognize the virus, and the immune tolerant microenvironment of the liver [8,9]. Therefore, novel therapeutic approaches to provide a finite treatment are required to improve care, and potentially increase functional hepatitis B virus (HBV) cure rates.
Major obstacles to achieving a functional HBV cure include the presence of covalently closed circular DNA (cccDNA) and exhausted, ineffective host immune responses [10]. Eradication of HBV could be achieved by therapeutic approaches targeting these viral and host factors that contribute to HBV persistence [7,10]. Eradication of HBV depends on both innate and adaptive immune responses, and the pathogen-recognition receptors are a potential therapeutic target [7]. The toll-like receptor-7 (TLR-7) expressed on plasmacytoid dendritic cells (pDC) and B cells is one such target [11]. In pDC cells, TLR-7 activation induces expression of type 1 IFNs, mainly IFN-α, and functional maturation of pDCs leading to increased antigen presentation, which in turn leads to activation of natural killer cells, dendritic cells, monocytes, B cells and cytotoxic CD8+ T cells [12,13]. In B cells, activation of TLR-7 induces maturation and secretion of antibodies, potentially promoting HBsAg loss and seroconversion [14,15].
JNJ-64794964 (JNJ-4964; formerly AL-034; or TQ-A3334) is a novel, oral and selective toll-like receptor-7 agonist. JNJ-4964 had potent anti-HBV activity in vitro, inducing antiviral IFNs, pro-inflammatory and anti-inflammatory cytokines and chemokines in human whole blood cells and peripheral blood mononuclear cells [16] and HBsAg seroconversion in preclinical studies [17].
The primary objectives of this first-in-human study (NCT03285620, AL-034-1201) were to assess the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of increasing single-ascending does (SAD) and multiple ascending doses of orally administered JNJ-4964 in healthy adults. The secondary objectives were to evaluate the effect of food on JNJ-4964 PK at a single dose level and the relationship between PK and safety.
Methods
Participants
Eligible participants were healthy adults 18–55 years old with a body mass index 18.0–30.0 kg/m2 (Supplementary Table 1).
Ethics approval
This study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practices and applicable regulatory requirements. The study protocol was reviewed by an independent Ethics Committee or Institutional Review Board. Participants provided their informed written consent to participate in the study.
Study design
This Phase I, randomized, double-blind, placebo-controlled study comprised two parts. JNJ-4964 starting dose was selected based on the minimum anticipated biological effect level (MABEL) approach in preclinical species (Supplementary appendix). In the SAD phase, single oral doses of JNJ-4964 were escalated, by cohort, under fasted conditions, and one cohort received the same dose in a fed state. In the subsequent phase of the study, dose and treatment regimen (every week [QW] versus every 2 weeks) was determined on the basis of safety, tolerability, and PK data from the SAD phase, as well as PK modeling and preclinical animal data (Janssen data on file). It was planned to include at least one cohort and two optional cohorts of multiple ascending doses.
For the SAD phase, 8–10 participants randomized per cohort received a single oral dose of JNJ-4964 0.2 (N = 6), 0.6 (N = 6), 1.25 (N = 8) or 1.8 mg (N = 6) or placebo (N = 2/dose cohort) under fasted conditions. Following a ≥6 week washout period, participants receiving 1.25 mg under fasted conditions were given 1.25 mg (N = 7) or placebo (N = 1) under fed conditions (a standard meal consumed before dosing) (Supplementary figure 1). The 1.25 mg cohort fasted and fed included a minimum of three females. The number of female participants was not pre-specified per protocol.
For the second phase of the study, only one cohort of eight participants received doses of 1.25 mg JNJ-4964 (N = 6) or matching placebo (N = 2) in fasted conditions in a QW regimen for 4 weeks (Supplementary figure 1).
Assessments
Eligible participants attended the clinic for a screening evaluation visit (within 28 days of dosing on day 1), baseline safety assessments (day–2), and study drug administration. For the SAD phase, participants were dosed (day 1) and remained in the clinic for 7 days for PK, safety, tolerability, and PD assessments. In the multiple dose (MD) phase, participants received study drug QW from day 1 and underwent study assessments in the clinic for 96 h after each dose. In both phases, participants returned to the clinic for weekly follow-up visits for 4 weeks after discharge.
Safety and tolerability were continuously assessed throughout the study. All adverse events (AEs) were recorded from the time of consent to the completion visit and were graded using the Division of AIDS toxicity grading scale [18]. At all scheduled study visits (per protocol) and at unscheduled visits (for evaluation of AEs), participants underwent physical examinations, electrocardiograms, vital sign checks, and clinical laboratory parameters (chemistry, hematology, and urinalysis). Ophthalmoscopy was performed at baseline and 2 weeks post last dose in the MD phase.
For PK assessments, following a single dose of JNJ-4964, intensive blood sampling was performed over the first 24 h, then daily through day 6. Weekly samples were collected through day 35. For weekly dosing of JNJ-4964 in the MD phase, blood samples were collected at baseline and post the first and fourth doses on days 1 (0.5, 1, 2, and 12 h) 2, and 4, and post the second and third doses on days 1 (0.5 h) and 4, and at follow-up week 2 or at an early withdrawal visit.
Urine samples for PK analysis were also collected in male participants. Plasma and urine concentrations of JNJ-4964 were evaluated using a validated liquid chromatography–tandem mass spectrometry method; the lower limit of quantification [LLOQ] was 0.75 pg/mL in plasma and 1 pg/mL in urine. PK parameters were estimated using non-compartmental analysis (WinNonlin®, Certara, Princeton, NJ, USA).
For PD assessments in the SAD phase, serum samples were collected at baseline and post-dosing on days 1 (1.5, 6, and 12 h), 2, 3, 5, and at follow-up day 14. In the MD phase, serum samples were collected at baseline and post the first and fourth dose on days 1 (2, 4, and 12 h), 2, 3, 5, and at follow-up day 7; and post the second and third doses on day 1 (12 h) and day 3.
IFN-α was assessed by an enzyme-linked immunosorbent assay (LLOQ = 12.5 pg/mL); 13 other cytokines (granulocyte colony-stimulating factor [GCSF]), granulocyte macrophage colony-stimulating factor [GMCSF], IFN-γ, IFN-γ-induced protein 10 [IP-10], interleukin [IL] 1ß, IL-1 receptor antagonist [IL-1 RA], IL-10, IL-12 subunit P40, IL-6, IL-8, macrophage inflammatory protein 1ß [MIP-1ß], monocyte chemotactic protein 1 [MCP-1], and tumor necrosis factor alpha [TNF-α]) were analyzed by the appropriate Luminex assays (LLOQ = 25 pg/mL). IFN-stimulated gene (ISG) expression analyses were conducted for ISG15, MX1, and OAS1, using quantitative polymerase chain reaction.
Statistical analyses
Overall, between 50 and 74 healthy participants were planned, which is within the range recommended in the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.
Descriptive statistics were used to summarize the plasma concentrations and PK parameters of JNJ-4964. The impact of food on PK parameters was analyzed using geometric least square mean point estimates with 95% confidence interval (CI) from a mixed-effects model on log-transformed values with treatment as fixed effect and participant as random effect.
Descriptive statistics were used to summarize PD parameters over time, and relative fold changes from baseline values were calculated. Reported values below the assay LLOQ were imputed as LLOQ-1.
Results
Participant demographics and disposition
Baseline demographics.
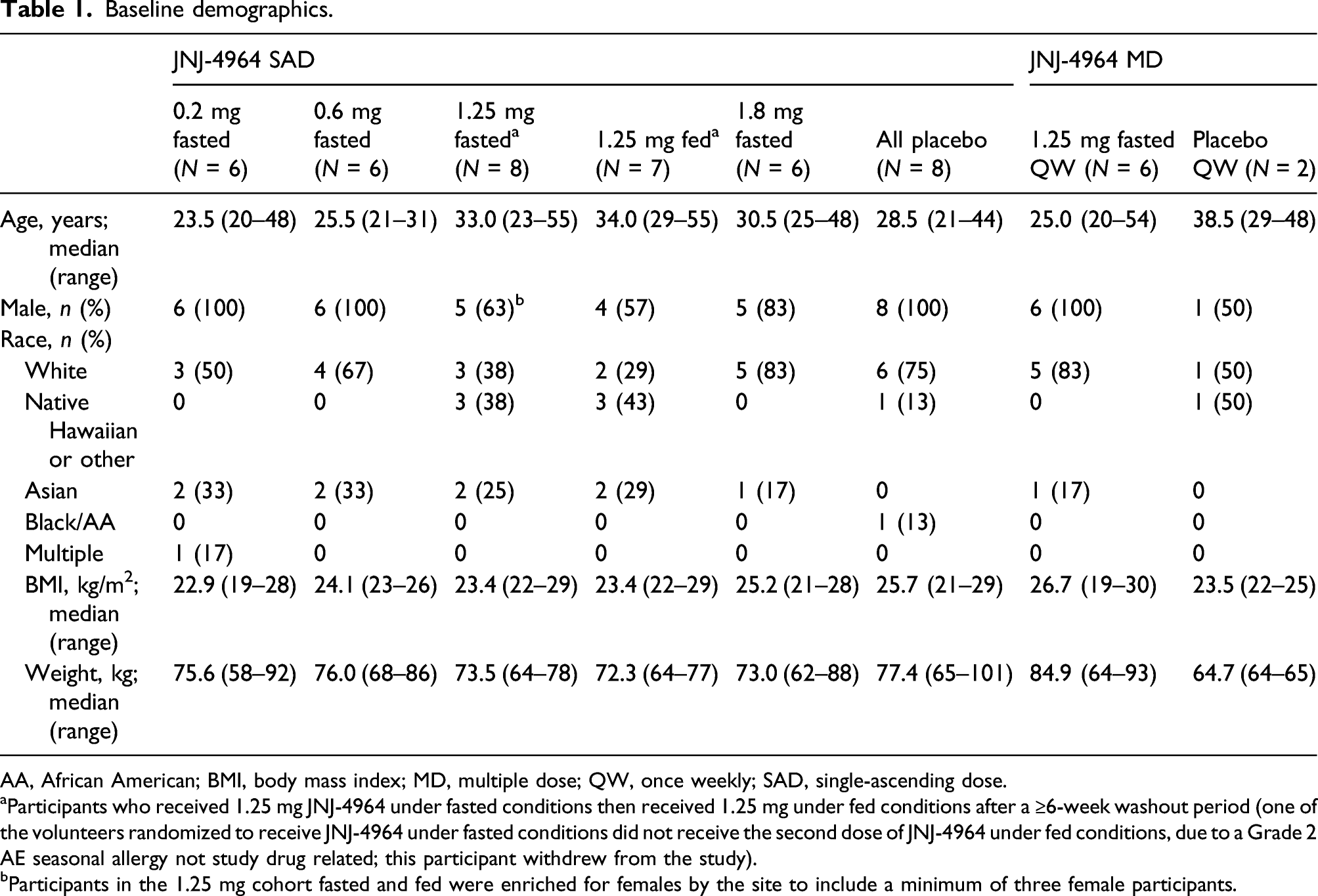
AA, African American; BMI, body mass index; MD, multiple dose; QW, once weekly; SAD, single-ascending dose.
aParticipants who received 1.25 mg JNJ-4964 under fasted conditions then received 1.25 mg under fed conditions after a ≥6-week washout period (one of the volunteers randomized to receive JNJ-4964 under fasted conditions did not receive the second dose of JNJ-4964 under fed conditions, due to a Grade 2 AE seasonal allergy not study drug related; this participant withdrew from the study).
bParticipants in the 1.25 mg cohort fasted and fed were enriched for females by the site to include a minimum of three female participants.
Thirty-two participants completed dosing and 31 completed the study (Supplementary Table 2). One participant receiving 0.2 mg JNJ-4964 withdrew their consent, although they had completed dosing. One participant receiving placebo withdrew their consent and did not roll-over into the food effect study, so did not complete dosing. One participant who received 1.25 mg JNJ-4964 under fasted conditions discontinued the study 17 days after dosing due to a Grade 2 AE of seasonal allergy considered not related to the study drug by the investigator. Dosing was not completed as the participant chose not to roll-over to receive 1.25 mg under fed conditions. There was a dose de-escalation for the 1.25 mg cohort from 1.8 mg to 1.25 mg after flu-like AEs (≤Grade 2) occurred in two participants in the 1.8 mg cohort.
In the MD phase, 20 participants were screened and eight were randomized and received 1.25 mg JNJ-4964 (N = 6) or matching placebo (N = 2) QW for 4 weeks under fasted conditions. Baseline demographics are shown in Table 1.
One participant who received 1.25 mg JNJ-4964 QW discontinued after the third dose due to an AE of ecchymosis (Grade 1), which was considered not related to the study drug by the investigator (Supplementary Table 2).
JNJ-4964 safety
All tested JNJ-4964 doses were generally well-tolerated with no major safety concerns. In the study, no deaths, other serious AEs or Grade ≥3 AEs occurred.
Summary of treatment-emergent AEs.
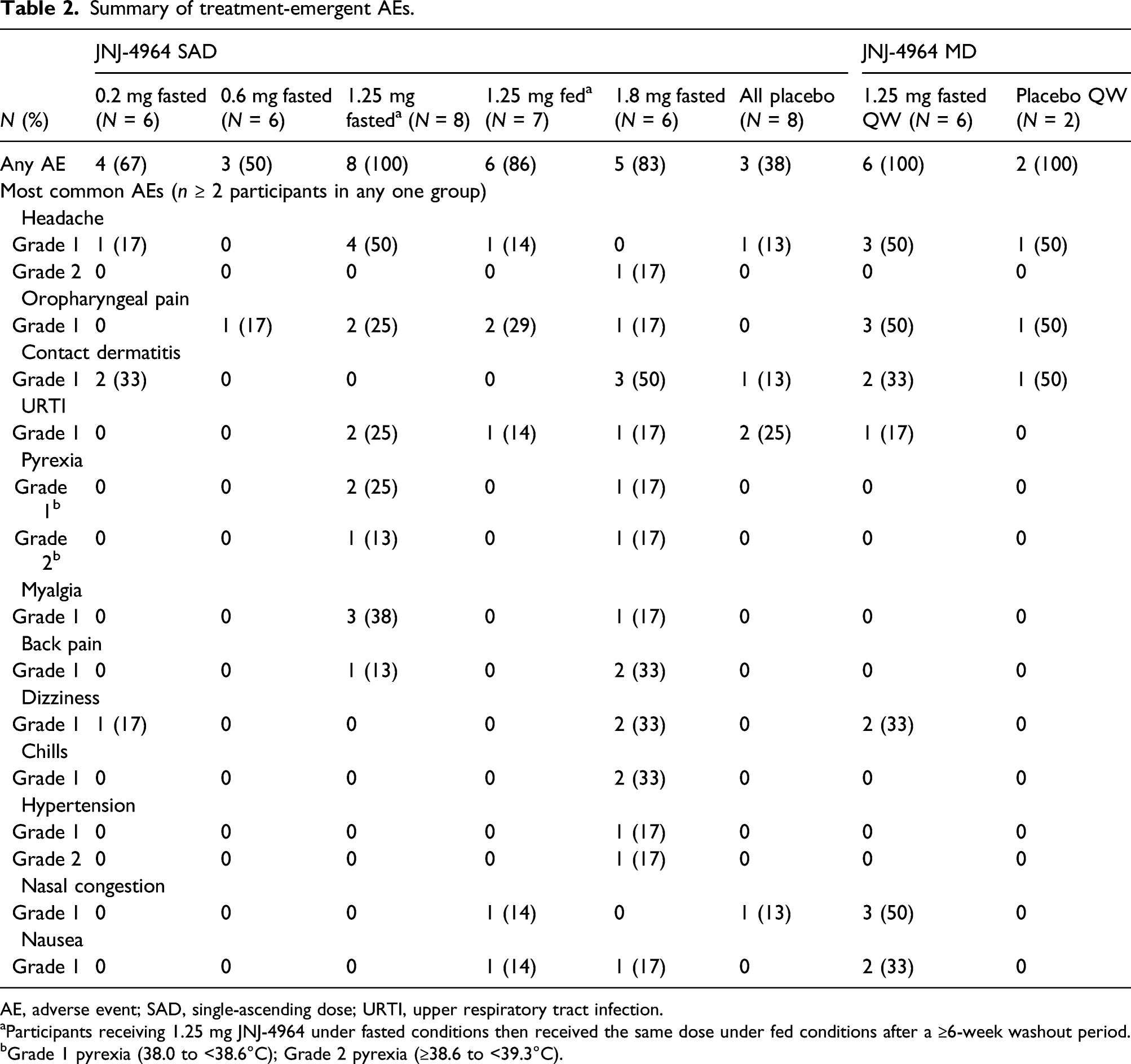
AE, adverse event; SAD, single-ascending dose; URTI, upper respiratory tract infection.
aParticipants receiving 1.25 mg JNJ-4964 under fasted conditions then received the same dose under fed conditions after a ≥6-week washout period.
bGrade 1 pyrexia (38.0 to <38.6°C); Grade 2 pyrexia (≥38.6 to <39.3°C).
Most AEs were not related to JNJ-4964. In 10/34 participants reporting AEs that were at least possibly related to JNJ-4964, these were most commonly (≥2 participants) pyrexia, headache, myalgia, oropharyngeal pain, hypertension, chills, and dizziness, which were all transient and reversible. Five participants (N = 3, 1.25 mg fasted; N = 2, 1.8 mg) had single events of fever and flu-like AEs between 24–48 h after dosing that resolved approximately 96 h after dosing. Treatment-related Grade 2 AEs occurred in three participants, one with pyrexia (1.25 mg fasted), one with pyrexia and headache, and one with hypertension (both in 1.8 mg cohort) (Table 2).
In the MD phase, the most common treatment-emergent AEs were headache, nasal congestion and oropharyngeal pain (Table 2).
Three out of six participants receiving 1.25 mg JNJ-4964 QW had an AE at least possibly related to JNJ-4964, including single events of retinal exudates (‘cotton wool spots’), nausea, arthralgia, headache, dyspnea, dry skin, and retching. No Grade 2 treatment-related AEs were reported.
A Grade 1 AE of bilateral retinal exudates was observed in one participant during a scheduled routine ophthalmologic examination 2 weeks after receiving the last 1.25 mg JNJ-4964 QW dose. Vision was not impaired, other ocular abnormalities were not detected, and the AE had resolved on a follow-up examination 6 weeks later. No history of hypertension, vasculitis, or diabetes was reported for this participant.
Most laboratory abnormalities in both phases were Grade 1 and none were reported as an AE.
In the SAD phase, three participants had Grade 4 laboratory abnormalities. One participant receiving placebo had elevated creatinine kinase levels. Two patients receiving 1.8 mg JNJ-4964 had low lymphocyte count (grade 4 <0.35 Giga/L) proximal to dosing (within 24 h), and both events resolved within 72 h. Three participants had Grade 3 laboratory abnormalities. One had increased triglycerides with 1.8 mg JNJ-4964 on day 1 improving to a Grade 1 abnormality on day 2. One participant had increased aspartate aminotransferase (AST) levels (placebo) and one had increased low-density lipoprotein (LDL)-cholesterol levels (1.25 mg JNJ-4964 fasted but also observed at screening), both in the follow-up period.
In the MD phase, Grade 3 and Grade 4 elevated creatinine kinase levels were observed, each in one participant receiving 1.25 mg JNJ-4964 QW. These changes were isolated elevations that occurred during the follow-up period after recent physical activity.
Other than the Grade 1 AE of retinal exudates (MD phase), the only clinically significant changes in physical examination, vital signs, or electrocardiograms was a Grade 1 erythema in the throat and cervical lymphadenopathy, considered probably treatment-related, which occurred in one participant in the 1.8 mg JNJ-4964 cohort (SAD phase). This participant also had a treatment-emergent Grade 4 low lymphocyte count.
JNJ-4964 pharmacokinetics
Summary of JNJ-4964 PK parameters.
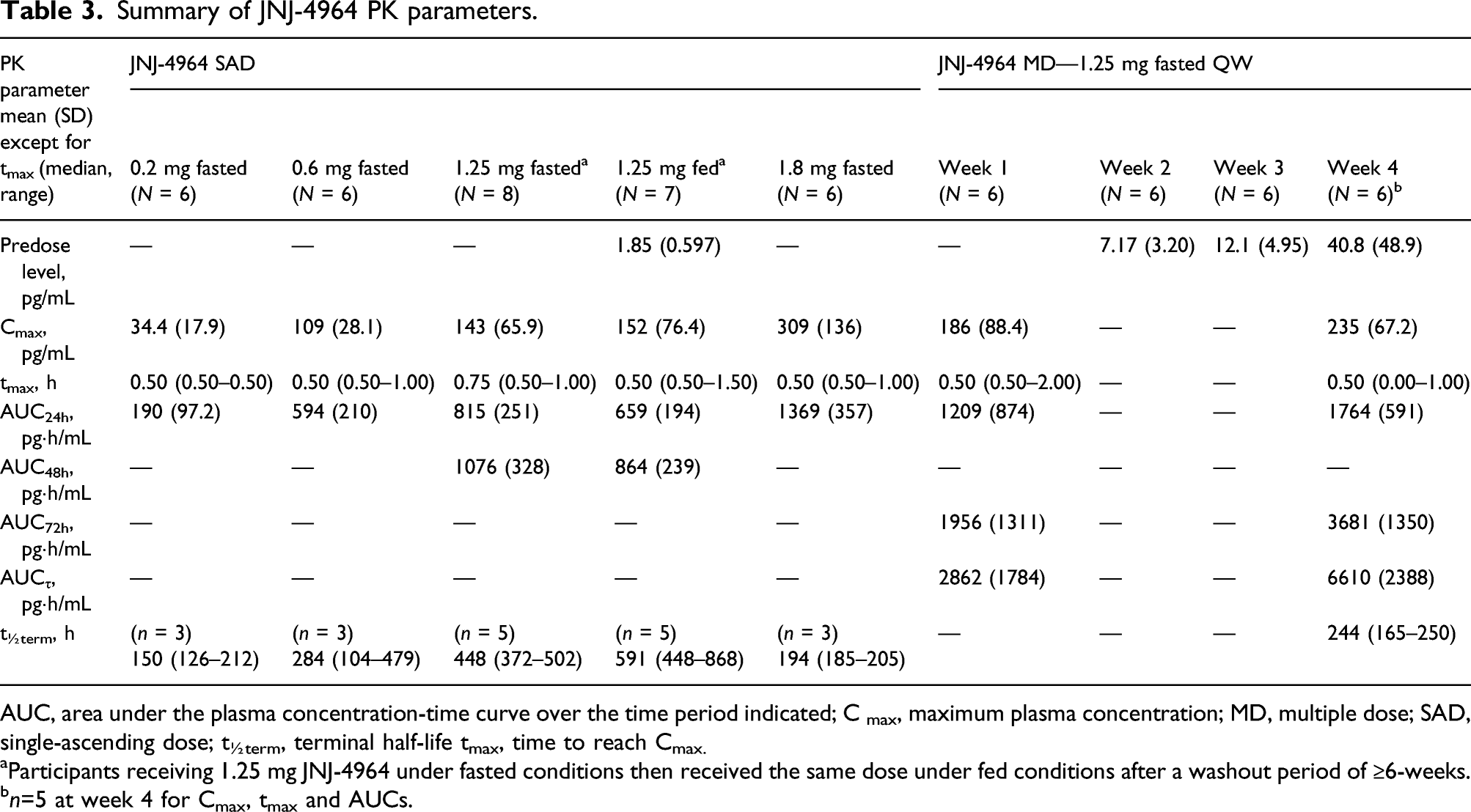
AUC, area under the plasma concentration-time curve over the time period indicated; C max, maximum plasma concentration; MD, multiple dose; SAD, single-ascending dose; t½term, terminal half-life tmax, time to reach Cmax.
aParticipants receiving 1.25 mg JNJ-4964 under fasted conditions then received the same dose under fed conditions after a washout period of ≥6-weeks.
bn=5 at week 4 for Cmax, tmax and AUCs.
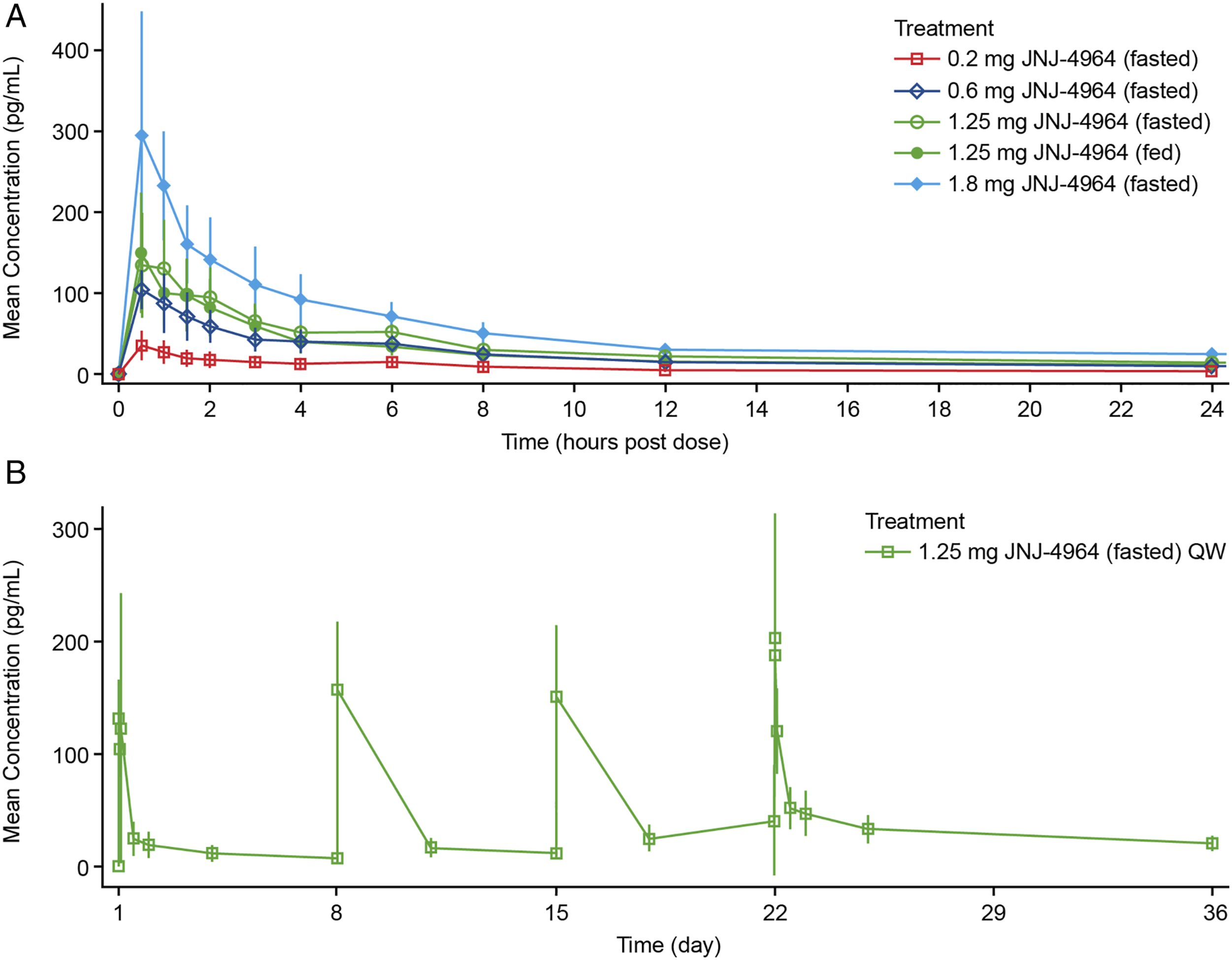
JNJ-4964 plasma concentrations (mean ± standard deviation) following single (
Following single oral JNJ-4964 doses of 0.2–1.8 mg (fasted), maximum JNJ-4964 plasma concentration (Cmax), and area under the plasma concentration-time curve from 0 to 24 h post-dose (AUC24h) increased dose proportionally (Table 3 and Figure 1A). Median tmax was similar across all JNJ-4964 cohorts (Table 3). For participants with an estimable apparent oral clearance, values were 149, 244 and 254 L/h after a single oral dose of 0.2, 0.6, and 1.25 mg of JNJ-4964 (fasted), respectively. Less than 0.2% of the JNJ-4964 dose was excreted as unchanged drug in the urine. After a single oral dose of 1.25 mg JNJ-4964 (fasted), mean renal clearance was 2.57 L/h for JNJ-4964.
Despite a washout period of ≥6 weeks following 1.25 mg JNJ-4964 (fasted), predose plasma concentrations (Cpredose) were quantifiable for all participants (mean [SD] Cpredose 1.85 [0.597] pg/mL) (Table 3). No significant differences were observed between JNJ-4964 PK parameters under fed versus fasted conditions (Supplementary Table 3). However, the 90% CIs were outside the 80.0–125.0% limits for bioequivalence. While the Cmax values under fasted and fed conditions were similar, AUC48h for JNJ-4964 was ∼17% lower for the fed versus fasted state, with a least square mean ratio point estimate of 83.45 (90% CI 65.89, 105.71) (Supplementary Table 3).
With multiple dosing of JNJ-4964, Cpredose increased after each weekly dosing (Figure 1B and Table 3). An increase in plasma Cmax was observed after 4 weeks of dosing (235 pg/mL at week 4 vs 186 pg/mL at week 1). After the last dose on day 22, JNJ-4964 plasma concentrations were still quantifiable at the last sampling timepoint (336 h post-dose). Accumulation was observed after 4 weeks of QW dosing, with mean accumulation ratios of 1.83, 2.27, and 2.75 for AUC24h, AUC72h, and AUCτ, respectively. Less than 0.22% of the JNJ-4964 dose was excreted as unchanged drug in urine within the first 24 h after the first and second doses.
JNJ-4964 pharmacodynamics
Number of participants with a ≥twofold change from baseline in cytokine expression or ISG expression within 96 h post-dosing with JNJ-4964. a
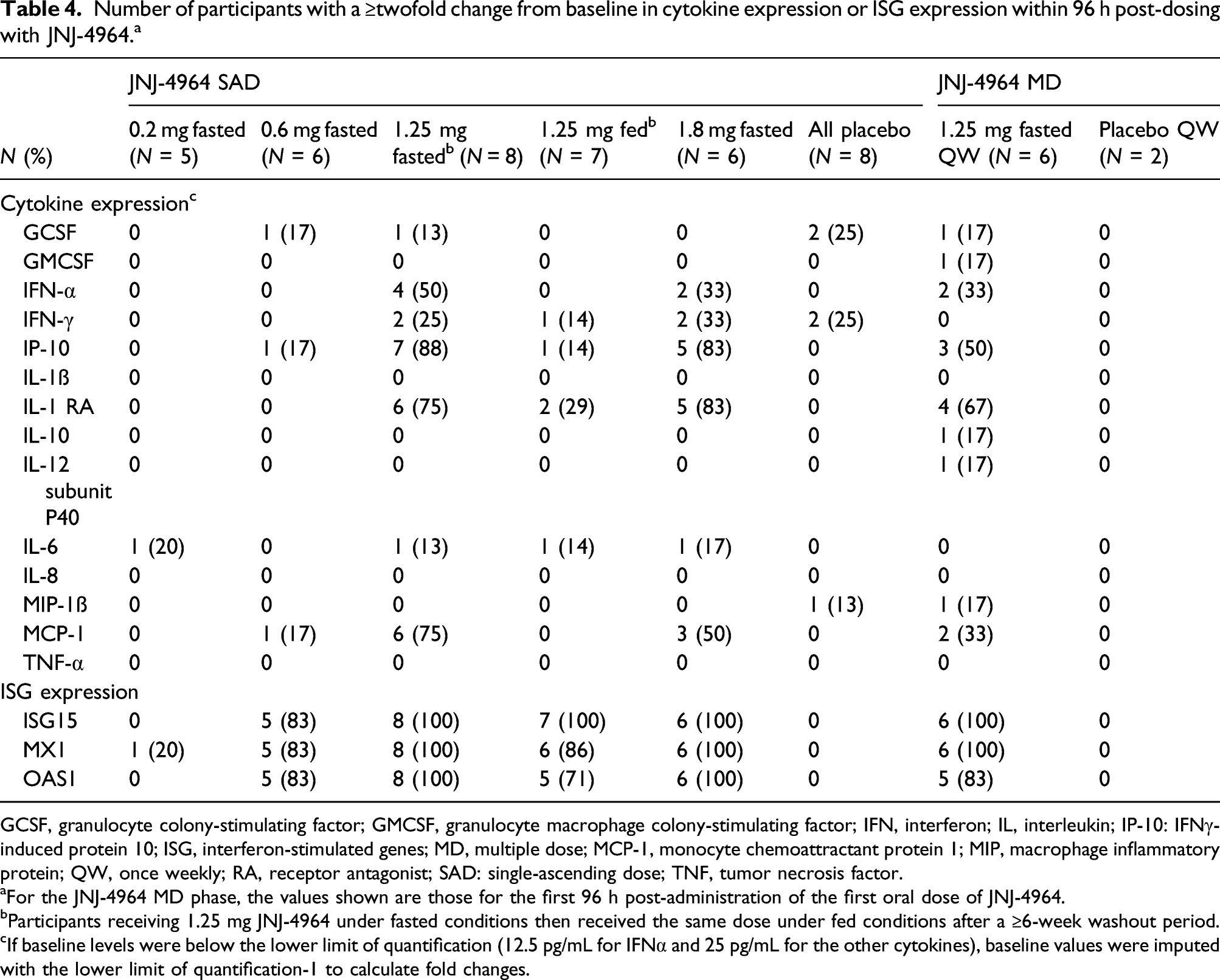
GCSF, granulocyte colony-stimulating factor; GMCSF, granulocyte macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; IP-10: IFNγ-induced protein 10; ISG, interferon-stimulated genes; MD, multiple dose; MCP-1, monocyte chemoattractant protein 1; MIP, macrophage inflammatory protein; QW, once weekly; RA, receptor antagonist; SAD: single-ascending dose; TNF, tumor necrosis factor.
aFor the JNJ-4964 MD phase, the values shown are those for the first 96 h post-administration of the first oral dose of JNJ-4964.
bParticipants receiving 1.25 mg JNJ-4964 under fasted conditions then received the same dose under fed conditions after a ≥6-week washout period.
cIf baseline levels were below the lower limit of quantification (12.5 pg/mL for IFNα and 25 pg/mL for the other cytokines), baseline values were imputed with the lower limit of quantification-1 to calculate fold changes.
Five of the six participants in the 0.6 mg JNJ-4694 group and all participants in the 1.25 mg fasted and 1.8 mg groups had a ≥twofold increase from baseline for all three ISGs within the first 96 h post-dosing, peaking within 12–24 h. No ISG induction was observed in the placebo group (Table 4).
In the 0.6 mg JNJ-4964 group, one out of the six participants showed ≥twofold increases from baseline of IP-10 and MCP-1. In the 1.25 mg fasted and 1.8 mg groups, levels of five cytokines (IFN-α, IP-10, IL-1 RA, MCP-1, and IL-6) increased ≥twofold from baseline within the first 96 h post-dosing but not with placebo (Table 4). Only increases in IFN-α, IP-10, IL-1 RA, and/or MCP-1, but not IL-6, were observed to be dose-dependent (Table 4 and Supplementary Figure 2).
Increased IFN-α expression (>LLOQ of 12.5 pg/mL) was only observed among participants who also had an increased expression in IP-10, MCP-1, and/or IL-1 RA in the 1.25 fasted and 1.8 mg groups (data not shown). In the 1.25 mg fasted and 1.8 mg groups, maximum concentrations of IFN-α (137.5–815 pg/mL), IP-10 (435.5–7186 pg/mL), IL-1 RA (80.7–5004 pg/mL), and MCP-1 (1065–13,055 pg/mL) were observed 12–24 h post-dosing. In the 1.25 mg fasted group, 2/3 females had a ≥twofold increase in IFN-α compared with 2/5 male participants. In the 1.8 mg group, the single female had a ≥twofold increase in IFN-α versus 1/5 male participants.
Cytokine levels (Supplementary Figure 2) and ISG expression (data not shown) returned to baseline levels within 48–96 h post-administration of a single oral dose of JNJ-4964. Increased expression of cytokines and ISGs was less frequently observed in the 1.25 mg fed state than in the fasted state (Table 4).
With multiple dosing of 1.25 mg JNJ-4964, increases were observed in the same cytokines and ISGs that were elevated in the single dosing 1.25 mg fasted group (Table 4). Neither enhanced nor tachyphylaxis (i.e., reduction) in cytokine and ISG induction responses were seen after the last dose compared with the first JNJ-4964 dose. For participants with increased IFN-α or IP-10 cytokine levels, values returned to baseline before administration of the subsequent dose.
Relationships between PD and safety parameters
With single JNJ-4964 dosing, in 3/8 participants in the 1.25 mg JNJ-4964 group and two of six participants in the 1.8 mg group, a positive relationship was observed between IFN-α levels ≥100 pg/mL and transient appearance of flu-like ≤Grade 2 AEs and reduction in lymphocyte count (Supplementary Figure 3).
In the MD phase, for the participant with “cotton wool spots,” IFN-α levels were <100 pg/mL at each timepoint, with a maximum of 97 pg/mL at 12 h after the second administration, and flu-like AEs (i.e., nausea and headache) were not observed.
Discussion
This first-in-human study investigated the safety, tolerability, PK, and PD of orally administered JNJ-4964 SAD and MD in healthy participants. JNJ-4964 at single doses up to 1.8 mg (fasted) and at multiple 1.25 mg QW doses for 4 weeks was generally well-tolerated and exhibited dose-proportional pharmacokinetics and dose-dependent, transient induction of certain cytokines and ISGs.
JNJ-4964 had an acceptable tolerability profile, which is an important requirement for chronic HBV treatments in order to facilitate treatment compliance. In both SAD and MD phases, no deaths, serious AEs, or ≥Grade 3 AEs were observed. In the SAD phase, two Grade 4 treatment-emergent laboratory abnormalities of low lymphocyte count and one Grade 3 event of increased triglycerides observed with 1.8 mg JNJ-4964 administration all resolved within 72 h. One Grade 3 increased LDL-cholesterol (1.25 mg JNJ-4964) in the SAD phase and one Grade 4 and one Grade 3 creatine kinase increase (1.25 mg JNJ-4964 QW) in the MD phase were observed in the follow-up periods. However, none of the observed treatment-emergent laboratory abnormalities were reported as an AE and were explained by reasons unrelated to study drug. These findings are consistent with a similar JNJ-4964 SAD study of 0.2–1.8 mg JNJ-4964 in 32 healthy Chinese participants [19], in which JNJ-4964 was generally well-tolerated, with no serious AEs and the majority of AEs were mild-to-moderate (≤Grade 2). The Grade 3 AEs were transient lymphopenia and neutropenia, which occurred in three female participants proximal to dosing, suggestive of JNJ-4964 on-target effects.
While an AE of ‘cotton wool spots’ considered possibly related to JNJ-4964 was observed in one participant during routine procedure 2 weeks after receiving the last JNJ-4964 dose in the MD phase, this event was asymptomatic and had resolved 6 weeks after observation. Cotton wool spots are thought to be axoplasmic debris caused by obstructions to axoplasmic flow in retinal ganglion cell axons due to mechanical or vascular causes [20,21], and they usually resolve within 6–12 weeks [22]. Cotton wool spots are commonly associated with diabetes mellitus, systemic hypertension or ischemic, embolic, connective tissue, neoplastic and infectious etiologies. In some cases, no underlying cause can be found [23]. However, this participant had no history of hypertension, vasculitis, or diabetes and did not have any low lymphocyte count abnormalities, but did experience flu-like AEs (i.e., nausea and headache) and increased IFN-α levels. Cotton wool spots have previously been observed in patients receiving interferon therapy for viral hepatitis and are usually asymptomatic and reversible [24–26].
A starting dose of 0.2 mg JNJ-4964 was chosen in the SAD phase, and escalation to 1.8 mg followed by de-escalation in subsequent participants to 1.25 mg JNJ-4964 was based on safety, tolerability, and PK data. JNJ-4964 was rapidly absorbed (tmax 0.5–0.75 h), followed by a rapid distribution phase, with a long t½term. Our JNJ-4964 PK results were consistent with those observed in healthy Chinese participants [19], in which JNJ-4964 exposure increased nearly proportionally with the dose, and tmax was reached at 0.5–0.6 h after drug administration, with moderate-to-slow elimination. Overall, no significant differences were observed between the JNJ-4964 PK parameters of the fed and fasted patients. As JNJ-4964 is not completely eliminated before the next dose (mean Cpredose 1.85 pg/mL), the decrease in exposure in the fed state may be underestimated. Based on PK, safety, tolerability, and PD data from the SAD phase, a starting dose of 1.25 mg QW JNJ-4964 mg in fasted conditions was selected for the MD phase.
Single oral doses of 1.25 and 1.8 mg JNJ-4964 dose-dependently and transiently induced cytokines including IFN-α, IP-10, IL-1 RA, and/or MCP-1, and all three ISGs (ISG15, MX1, and OAS1) in serum. The results are consistent with the other JNJ-4964 SAD study [19], in which all JNJ-4964 doses except 0.2 mg induced expression of these cytokines and ISGs between 12–72 h post-dose and the effect was dose-dependent. TLR-7 agonists, such as JNJ-4964, targeted to the liver could be beneficial for CHB treatment by potentially modulating the immune system with an acceptable safety profile. JNJ-4964 induction of IFN-α and other cytokines to detectable levels in the plasma suggests TLR-7 activation on pDC cells and a subsequent antiviral immune response, as a result of functional maturation of pDCs, leading to increased antigen presentation, and activation of natural killer cells, dendritic cells, monocytes, B cells, and cytotoxic CD8+ T cells [12,13].
An increase in the same cytokines and ISGs was also observed in the JNJ-4964 1.25 mg QW group, with no evidence for enhanced induction of PD responses or tachyphylaxis after the last dose compared with the first dose of JNJ-4964. Increases in IFN-α or IP-10 cytokine levels returned to baseline before administration of the subsequent dose despite observed accumulation of JNJ-4964. Accumulation of JNJ-4964 is likely due to its slow elimination and not due to a change or increase in absorption, since Cmax was similar after repeated dosing. The remaining fraction of JNJ-4964 in the circulation may not be sufficient to elicit concerted type 1 IFN responses in pDC cells at the level of gut-associated lymphoid tissue, a proposed mechanism of TLR-7 activation [27]. Preclinical data with the TLR-7 agonist, vesatolimod, supports this assertion since systemic exposure to vesatolimod via the intravenous route generates a minimal PD response compared with exposure via the oral route [27].
In 3/8 participants in the 1.25 mg JNJ-4964 group and 2/6 participants in the 1.8 mg group, IFN-α levels ≥100 pg/mL coincided with transient appearance of flu-like ≤Grade 2 AEs and a low lymphocyte count, which is expected with increased IFN-α levels. These observations are consistent with the expected safety profile for TLR-7 agonists, which include transient flu-like AEs and lymphocyte decrease correlated with IFN-α exposure [28–33].
The present study has some limitations. As the study was conducted in healthy participants, PK, PD, and safety need to be confirmed in CHB patients. Also, participants were predominantly male and white. In the other JNJ-4964 SAD study [19], the ratio of male to female participants was 1:1. Importantly, the JNJ-4964 response in females, as indicated by transient Grade 3 lymphopenia and neutropenia, was greater than in males.
In conclusion, in healthy participants, the evaluated single oral doses of 0.2–1.8 mg JNJ-4964 and multiple oral doses of 1.25 mg JNJ-4964 were generally well-tolerated, with dose-proportional PK and induced cytokines and ISGs, with the potential of target engagement of this TLR-7 agonist.
Supplemental Material
sj-pdf-1-avt-10.1177_13596535211056581 – Supplemental Material for Safety, tolerability, pharmacokinetics, and pharmacodynamics of oral JNJ-64794964, a TLR-7 agonist, in healthy adults
Supplemental Material, sj-pdf-1-avt-10.1177_13596535211056581 for Safety, tolerability, pharmacokinetics, and pharmacodynamics of oral JNJ-64794964, a TLR-7 agonist, in healthy adults by Edward Gane, Mina Pastagia, Ullrich Schwertschlag, An De Creus, Christian Schwabe, Joris Vandenbossche, Leen Slaets, Bart Fevery, Ilham Smyej, Liviawati S Wu, Rui Li, Samia Siddiqui, Abbie Oey, Clark Musto and Pieter Van Remoortere in Antiviral Therapy
Footnotes
Acknowledgements
We express our gratitude to the participants in this study. We also thank other Janssen staff members for their contributions to this study, including Benno Ingelse, Janssen Pharmaceutica NV. This study was sponsored by Janssen BioPharma Inc. Medical writing support for the development of this article, under the direction of the authors, was provided by Ian Woolveridge, MA (Hons), PhD, of Ashfield MedComms, an Ashfield Health company, and funded by Janssen.
Author contributions
EJG and CS contributed to the conduct of the study as investigators and to the interpretation of the data. MP, LS contributed to analysis and interpretation of the data. AdC, JV, BF, IS, LSW, SS, AO, CM, BB, and PVR contributed to the design of the study and analysis and interpretation of the data. US was an independent trial consultant and participated in the review and interpretation of the data. All authors were involved in the development of the primary manuscript, interpretation of data, and have read and approved the final version, and have met the criteria for authorship as established by the ICMJE.
Disclosures statement
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MP, AdC, JV, LS, BF, IS, LSW, SS, AO, CM, BB, and PVR are employees of Janssen Pharmaceuticals and may be Johnson & Johnson stockholders. US is an independent trial consultant. EJG has been an advisor and/or speaker for AbbVie, Gilead, Janssen, Novartis, Roche, and Merck. CS has advised for Johnson & Johnson and Vir Biotechnology.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Janssen BioPharma Inc
Author’s note
Part 1 (SAD) data were presented in part at EASL, The International Liver Congress, Vienna, Austria, April 10–14, 2019 (abstract and poster FRI-198).
Data availability statement
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at ![]() .
.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.