
Abstract
Background:
The objective of the RANGER II SFA long lesion cohort analysis was to evaluate the safety and effectiveness of the Ranger drug-coated balloon (DCB) in patients with lesion lengths greater than 100 mm.
Methods:
Patients from the RANGER II SFA randomized controlled trial and long balloon sub-study were included in the long lesion cohort if their baseline lesion measurement was > 100 mm and if they had been treated with a RANGER DCB. Patients had symptomatic lower limb peripheral artery disease and Rutherford classification 2–4 symptomatology. The endpoints of interest included the 12-month target lesion primary patency and freedom from major adverse events (MAEs).Additional patient outcomes including changes in Rutherford classification were also evaluated.
Results:
A total of 129 patients met the inclusion criteria and were included in the long lesion cohort. Mean lesion length was 144.5 ± 31.7 mm. Seventy-five lesions had Peripheral Arterial Calcium Scoring System (PACSS) grades 3 (33.3%, 43/129) and 4 (24.8%, 32/129). The Kaplan–Meier estimate of the primary patency rate at 12 months was 88.0%. The rate of freedom from MAEs at 12 months was 95.1% (117/123; 95% CI: 89.7%, 98.2%); all MAEs were clinically driven target lesion revascularization (4.9%, 6/123). The 12-month mortality rate was 2.4% (3/125).
Conclusions:
Patients with lesions > 100 mm treated with Ranger DCBs demonstrated excellent 1-year safety and efficacy results, comparable to those of the overall RANGER II SFA randomized clinical trial. This suggests that the Ranger DCB can provide consistent results regardless of lesion length.
Keywords
See commentary: Klein AJP. The safety and efficacy of ‘going long’. Vasc Med 2022;27:467–469.
Background
There are a variety of challenges associated with treatment of long lesions in the superficial femoral artery (SFA). Long lesions are more complex in terms of calcification, occlusion rate, and by definition, lesion length itself. All three of these characteristics have been shown to be associated with decreased patency rates over time.1,2 Lesion length > 100 mm and occlusion status are the main criteria used for the TransAtlantic Inter-Society Consensus (TASC) II categorization of femoral popliteal lesions because of their historical association with endovascular versus surgical treatment outcomes. 2 In current clinical settings, long lesions may require a combination of different treatment techniques to maximize and maintain patency, which must be coupled with close surveillance. Revascularization strategies may include percutaneous transluminal angioplasty (PTA), atherectomy, and stent deployment, among others. Increasing lesion length is associated with more frequent use of bail-out stenting and increased risk of the need for target lesion revascularization (TLR).3–6 Collectively, these factors help to explain the association between longer lesions and steep loss of patency observed beyond 1 year following drug-coated balloon (DCB) treatment.5,7,8
The use of paclitaxel-containing devices has been shown to improve duration of patency with a reduction in interventions for maintenance as compared to uncoated devices in the endovascular treatment of femoropopliteal lesions. Improvement attributed to DCB treatment relative to nondrug therapy has been demonstrated across a range of lesion lengths, including SFA lesions longer than 100 mm.4–6,9
Several DCBs for peripheral artery use are available and differ in terms of drug dose, type of excipient, and other characteristics that affect the release kinetics and, in turn, the efficiency of drug transfer to target tissue, tissue drug levels, and intraprocedural drug loss. The Ranger Paclitaxel-Coated PTA Balloon Catheter (Boston Scientific, Marlborough, MA, USA) is a low dose DCB with a dose density of 2 µg/mm2. In animal models, the Ranger DCB has demonstrated effective drug transfer into arterial tissue10,11 and neointimal inhibition, 12 as well as lower downstream tissue drug levels than other DCBs.10,11 The recently published RANGER II SFA randomized controlled trial (RCT) demonstrated that the Ranger DCB had significantly better effectiveness than standard PTA through 1 year of follow-up, with a good safety profile. 13
The primary objective of the RANGER II SFA long lesion cohort study was to utilize data collected in the globally conducted RANGER II SFA randomized trial and the associated long balloon sub-study (LBSS) to assess the effectiveness and safety of the Ranger DCB in the treatment of lesions 100 mm or longer located in the SFA and/or the proximal popliteal artery.
Methods
RANGER II SFA RCT and the Long Balloon Sub-study
The RANGER II SFA randomized controlled trial (RCT) (ClinicalTrials.gov Identifier: NCT03064126) is a prospective, multicenter, randomized, controlled, single blind superiority study (RCT) that included patients from 67 sites across six countries. 13 Balloon lengths available for the RCT were 30, 40, 60, 80, and 100 mm. A concurrent LBSS was conducted utilizing DCB lengths 120, 150, and 200 mm at select sites in Europe and New Zealand.
The ‘long lesion’ cohort included patients from the main RCT and those from the LBSS who received index treatment for the target lesion with the Ranger DCB and had a target lesion length of 100 mm or greater by core laboratory angiographic assessment (Figure 1). These patients were pooled to create the cohort presented here.
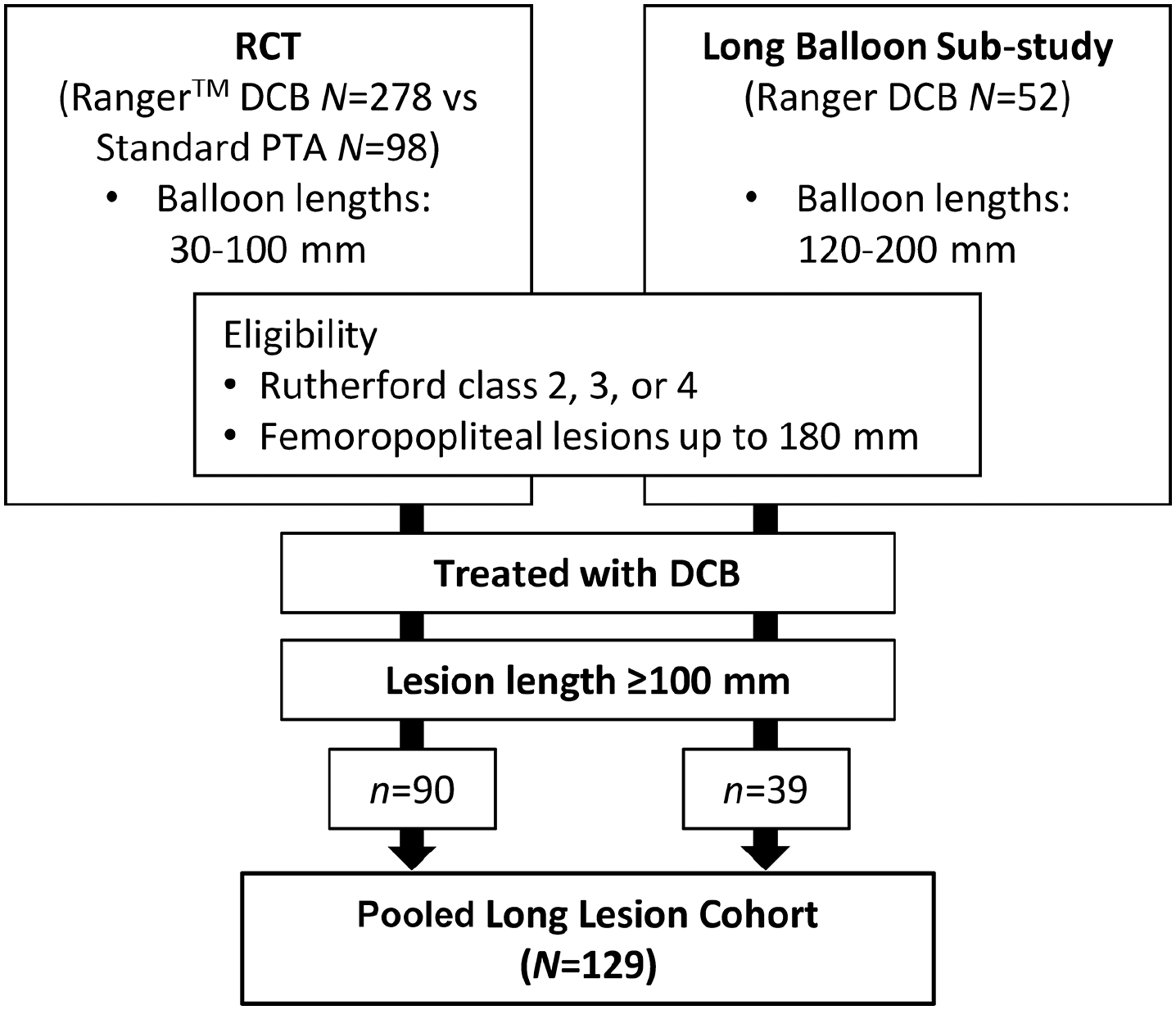
RANGER II superficial femoral artery (SFA) long lesion patient cohort. The pooled long lesion cohort comprises patients from the RANGER II SFA RCT and long balloon sub-study who were treated with the DCB and who had a lesion length ⩾ 100 mm.
The study protocol for the RANGER II SFA trial was approved (or permission to conduct the trial was granted) by each study site’s respective institutional review board (IRB), ethics committee (EC), or research ethic board; the LBSS protocol was approved by IRBs and ECs in Europe and New Zealand only. The study was conducted with ISO 14155 and principles consistent with the Declaration of Helsinki.
Eligibility criteria
Eligibility criteria for the RCT and sub-study were identical except for maximum occlusion length, as described below. Potential study subjects provided informed consent before beginning study-specific tests and procedures. Briefly, inclusion criteria were: having chronic symptomatic lower limb ischemia (Rutherford classification of 2, 3, or 4) and having a target lesion in the native SFA and/or proximal popliteal artery to the P1 segment (reference vessel diameter between 4 mm and 8 mm (inclusive) by visual estimate, minimum stenosis ⩾ 70%). 14 Successful predilatation without major dissection was required prior to enrollment. Tandem lesions were eligible provided that they were: (1) separated by a gap of ⩽ 30 mm; (2) total combined lesion length, including gap, meets angiographic requirements with total combined length ⩽ 180 mm; and (3) able to be treated as a single lesion. Patent popliteal and infrapopliteal arteries were required, with at least one of three vessels patent with < 50% stenosis to the ankle or foot. Both studies allowed for total lesion length up to 180 mm by visual estimate; the LBSS allowed occlusion up to 150 mm, whereas ⩽ 100 mm was allowed in the RCT.
Exclusion criteria included: chronic renal insufficiency with serum creatinine > 2.0 mg/dL within 30 days of index procedure or treatment with dialysis; failure to successfully cross the target lesion with a guidewire and successfully predilate the target vessel; presence of severe calcification rendering the lesion undilatable; or use of adjunctive primary treatment modalities (e.g., laser, atherectomy, scoring/cutting balloon, other debulking devices) during the index procedure. Treatment of the target lesion with atherectomy or DCB in the past 12 months were grounds for study exclusion, as were having ever been treated with surgery or a stent. If the lesion was restenotic, the most recent PTA treatment must have been more than 3 months prior to enrollment. Patients had to be free of significant inflow disease which could not be treated prior to the target lesion treatment. Additional details regarding complete inclusion and exclusion criteria have been previously published. 13
The site investigator’s imaging assessment was used to determine angiographic subject eligibility for the trial; independent analyses from the Angiographic Core Laboratory conducted by the Harvard Medical Faculty Physicians at Beth Israel Deaconess Medical Center (Boston, MA, USA) were used for final data analyses. Data collected and assessed by the core lab included angiograms from the index procedure and any subsequent revascularization procedure up to 12 months postindex procedure.
Randomization and masking
For the main RCT, treatments were randomly assigned after the patient was deemed to meet the eligibility criteria and the target lesion was both successfully crossed with a guidewire and predilated. The single-arm LBSS was nonrandomized with all subjects receiving the Ranger DCB. Additional details regarding randomization and masking in RANGER II have been previously published. 13
Procedures
Procedure descriptions were previously published. 13 Briefly, the Ranger DCB was used in accordance with the directions for use included with the device. Patients were allowed to have been treated with one or more overlapping balloons to treat the total lesion length.
Successful predilatation was required before DCB use. Per the instructions for use, the diameter of the predilatation balloon was smaller than that of the DCB to be used. The DCB was inflated for 3 minutes per the directions for use; postdilatation was performed at each investigator’s discretion. Bare metal stents were permissible to treat peri-procedural dissections or, in the case of residual stenosis (⩾ 50%) in patients in whom adequate results were not obtained, with prolonged low-pressure postdilatation.
Minimum requirements for anticoagulant and antiplatelet administration were applied to all study groups. Anticoagulant administration prior to and during the procedure was consistent with current clinical practice and dual antiplatelet therapy was required for at least 30 days for patients who did not receive a stent and at least 90 days for those who did receive a stent. Antiplatelet monotherapy was recommended throughout trial participation.
Clinical follow-up
All participants had scheduled clinical follow-up visits at 1-, 6-, and 12-months postindex procedure. Data collected at these appointments included ankle–brachial index (ABI), Rutherford classification, and duplex ultrasonography, medication assessment and adverse events. Functional and quality of life outcome measures included the 6-minute walk test (6 and 12 months only), Walking Impairment Questionnaire (WIQ), and the EQ-5D health-related quality of life questionnaire for RCT patients only; these data were not collected for the LBSS. RCT patients will be followed up through 5 years postindex treatment; patients in the LBSS were only followed for 1 year.
Outcome definitions
The primary and secondary endpoints described apply to the original RCT and LBSS; analogous post hoc analyses were conducted for the long lesion pooled cohort outcomes presented here.
For the main RCT and the LBSS, the primary effectiveness endpoint was 12-month primary lesion patency, defined as having a core laboratory-assessed duplex ultrasound peak systolic velocity ratio ⩽ 2.4 in the absence of clinically driven TLR or bypass of the target lesion. Clinically driven TLR (CD-TLR) was defined as: (1) any reintervention at the target lesion postindex procedure due to recurrent symptoms; or (2) an ABI/toe–brachial index (TBI) decrease of > 0.15 or decreased ⩾ 20% as compared with the postprocedure baseline ABI/TBI in the treated segment; TBI was permitted in cases in which vessels were incompressible. Duplex ultrasound assessment was conducted by an independent core vascular ultrasound laboratory (VASCORE, Boston, MA, USA); TLR was adjudicated by the Clinical Event Committee (CEC).
Secondary outcomes for the RCT and LBSS included measures of technical and procedural success and clinical outcomes, including changes in Rutherford classification and hemodynamic improvement. Sustained clinical improvement was defined as improvement in Rutherford classification from preprocedure by at least one category without the need for TLR. Hemodynamic improvement was defined as achieving an ABI ⩾ 0.90 or improvement of ABI by ⩾ 0.1 as compared to the preprocedure value without the need for repeat revascularization.
The primary safety endpoint was major adverse events (MAEs), defined as all TLR, major amputations through 12 months, and all-cause death within 1 month of index procedure. The independent CEC adjudicated all deaths, TLR, target vessel revascularization (TVR), and target limb amputations throughout the duration of the study.
Statistical analyses
All enrolled patients from the main RCT and the single arm LBSS with lesion length of ⩾ 100 mm were included in the long lesion cohort analytic set. Estimates and 95% CIs were reported for the primary and secondary endpoints; no formal hypotheses were proposed for these endpoints. The Kaplan–Meier product-limit method was used to estimate the event or event-free rates for time-to-event endpoints, including time to primary patency and time to freedom from TLR. Kaplan–Meier patency estimates were based on the time-to-event of TLR and/or 12-month duplex ultrasound patency failure up to 395 days (i.e., full 1-year follow-up window). Statistical analyses were performed with SAS software, Version 9.2 or higher (SAS Institute Inc., Cary, NC, USA).
Results
Patients
Patients were enrolled in the RCT and LBSS between March 2017 and August 2018. The long lesion pooled cohort included 129 patients; of these, 90 (69.8%) were from the Ranger II SFA RCT. The patient flow for the pooled cohort is shown in Figure 1. Primary safety outcome data were available through 12 months for 95.3% of patients (123/129); primary effectiveness outcome data were available through 12 months for 87.5% (113/129) patients. Three patients died prior to completing the 12-month visit.
Baseline characteristics
Baseline and clinical characteristics for patients included in the long lesion cohort are detailed in Table 1. The majority of patients in this cohort had hypertension (82.9%, 107/129), and 34.1% (44/129) had a diagnosis of diabetes mellitus. Over half of these patients were previously smokers (55.8%, 72/129): 31.8% (41/129) reported being current smokers and 12.4% (16/129) reported no history of smoking. Mean lesion length was 144.5 ± 31.7 mm; median lesion length was 139.0 mm (range: 100.2, 267.8) (Table 2). Peripheral Arterial Calcium Scoring System (PACSS) grades 3 and 4 were 33.3% (43/129) and 24.8% (32/129), respectively. Of the 129 patients included, 42 (32.6%) had 100% occlusion.
Baseline demographic and clinical characteristics of the long lesion cohort. (N = 129).
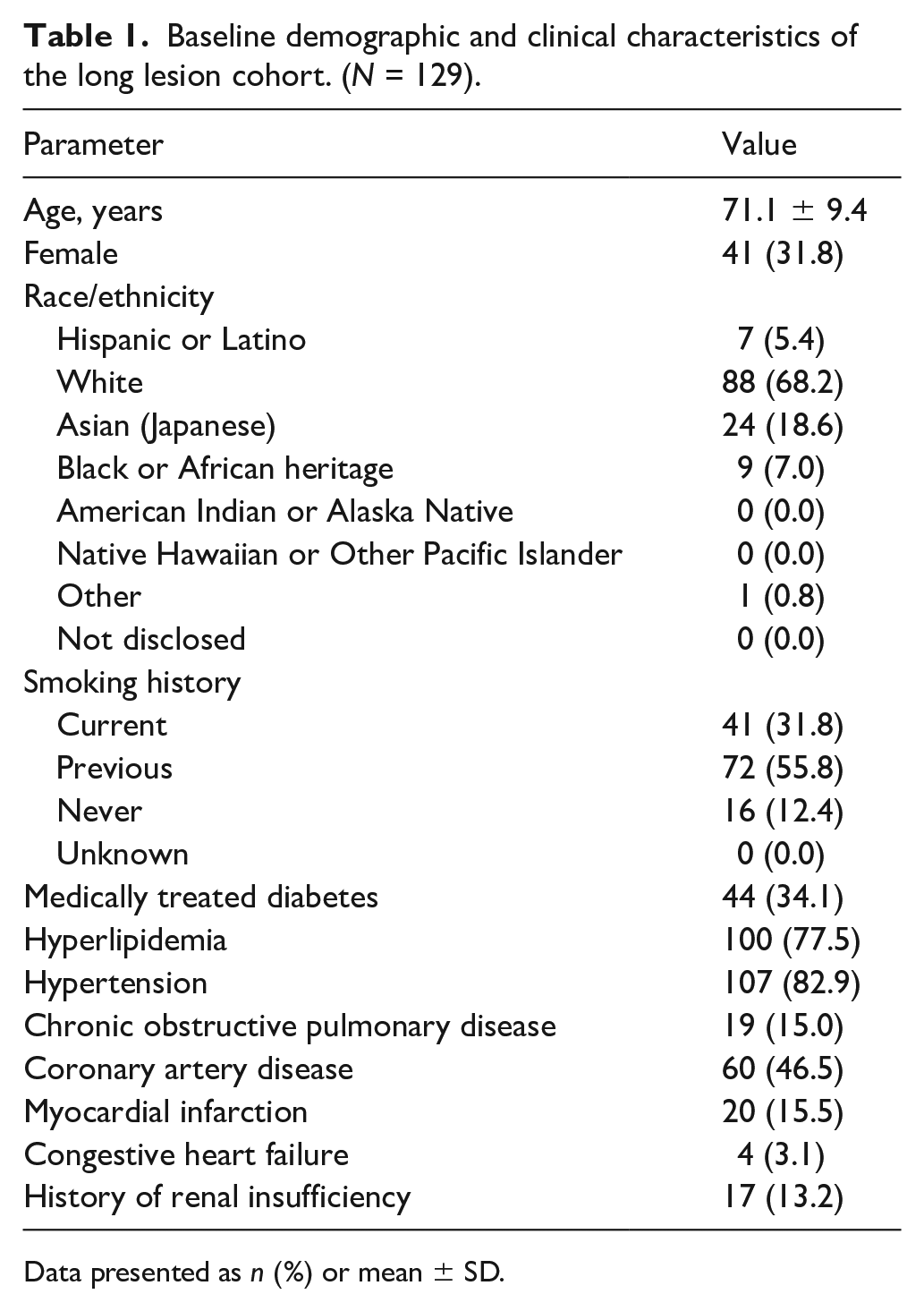
Data presented as n (%) or mean ± SD.
Preprocedure angiographic characteristics (core laboratory) (N = 129).
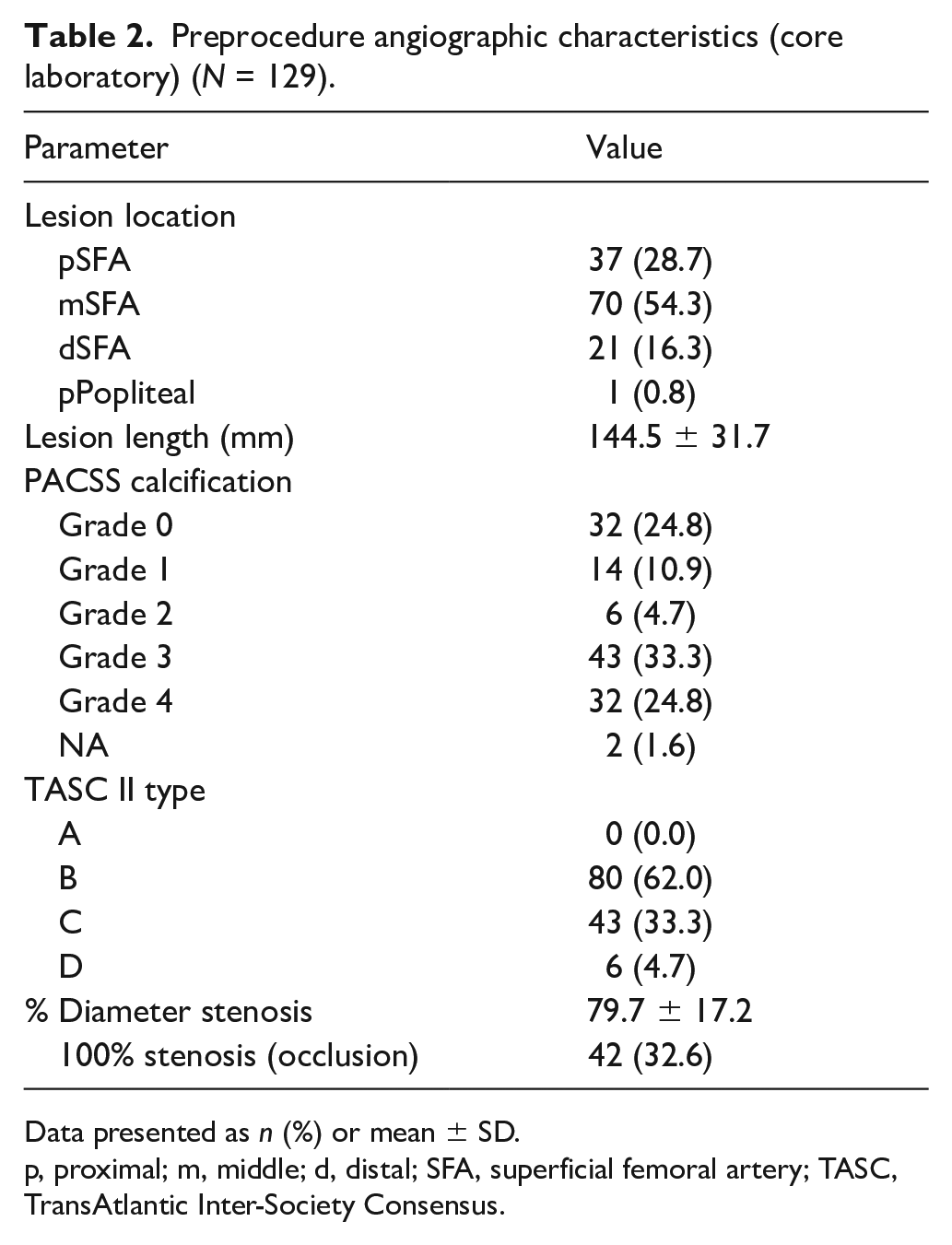
Data presented as n (%) or mean ± SD.
p, proximal; m, middle; d, distal; SFA, superficial femoral artery; TASC, TransAtlantic Inter-Society Consensus.
Predilatation was performed in all 129 patients; postdilatation was performed in 20.9% (27/129) of patients. Stent bail-out, all bare metal, was used in 7.0% (9/129) of patients. The mean diameter of the predilatation balloons was 4.6 mm (SD = 0.8), compared with 5.4 mm (SD = 0.8) for the Ranger DCB. The mean number of balloons used for predilatation was 1.2 (SD = 0.5) and 1.8 (SD = 0.6) for the Ranger DCB.
Primary effectiveness and safety outcomes
The Kaplan–Meier estimate of the primary patency rate at 365 days was 88.0% (Figure 2A). The safety endpoint of freedom from 12-month MAE was 95.1% (117/123; 95% CI: 89.7%, 98.2%); all MAE were CD-TLR (4.9%, 6/123; 95% CI: 1.8%, 10.3%; Table 3). The 12-month mortality rate was 2.4% (3/125). The three deaths were adjudicated by the Clinical Events Committee as: vascular (63 days postindex procedure, retroperitoneal hemorrhage subsequent to stent perforation of the iliac artery in the nontarget limb, unrelated to study treatment); cardiac (116 days postprocedure, myocardial infarction); and noncardiovascular (228 days postprocedure, sepsis). One patient was from the RCT, and this death was included in the 1-year mortality rate reported previously for that study. 13
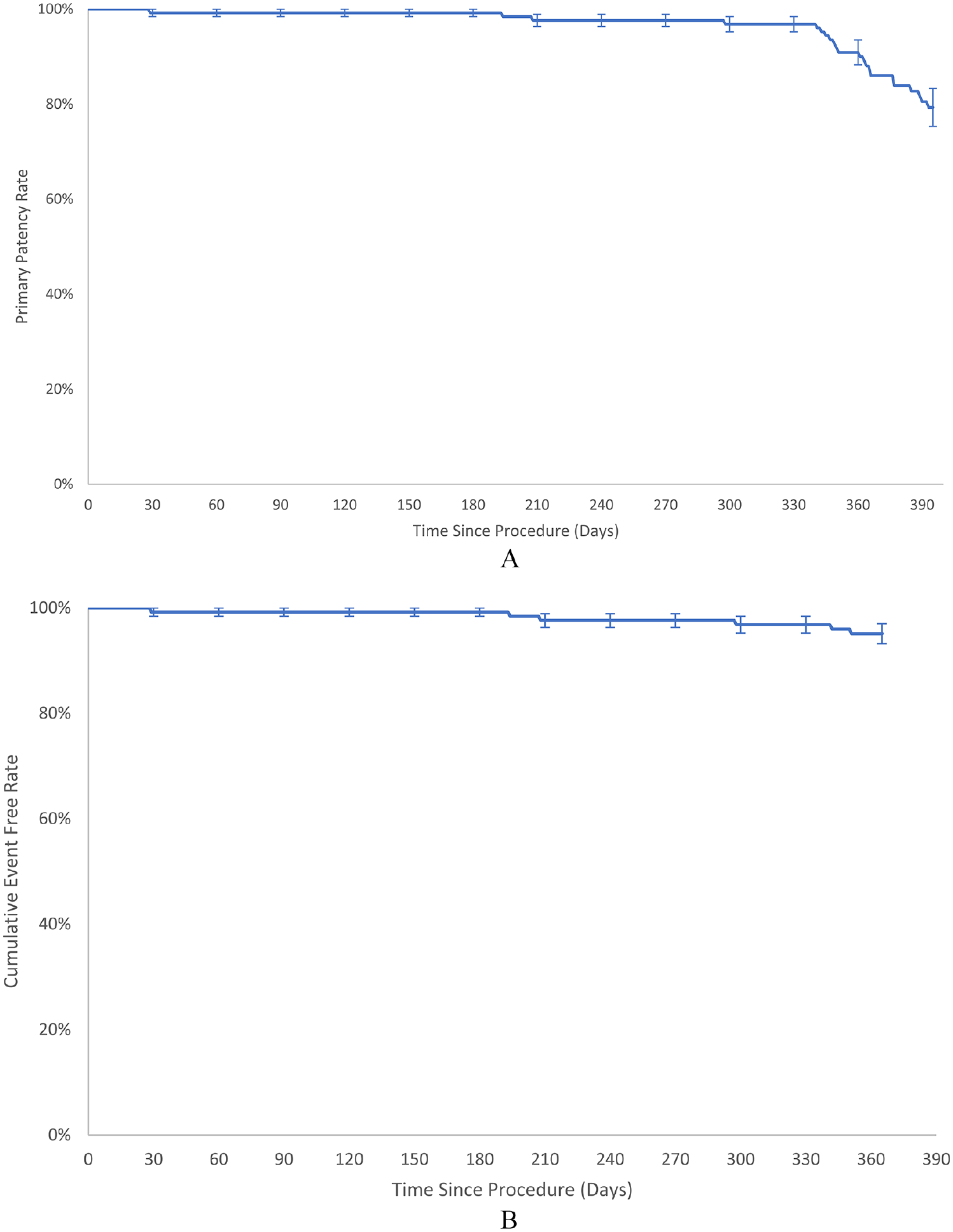
Kaplan–Meier curves of
Clinical outcomes at 12 months after drug-coated balloon treatment (N = 129).
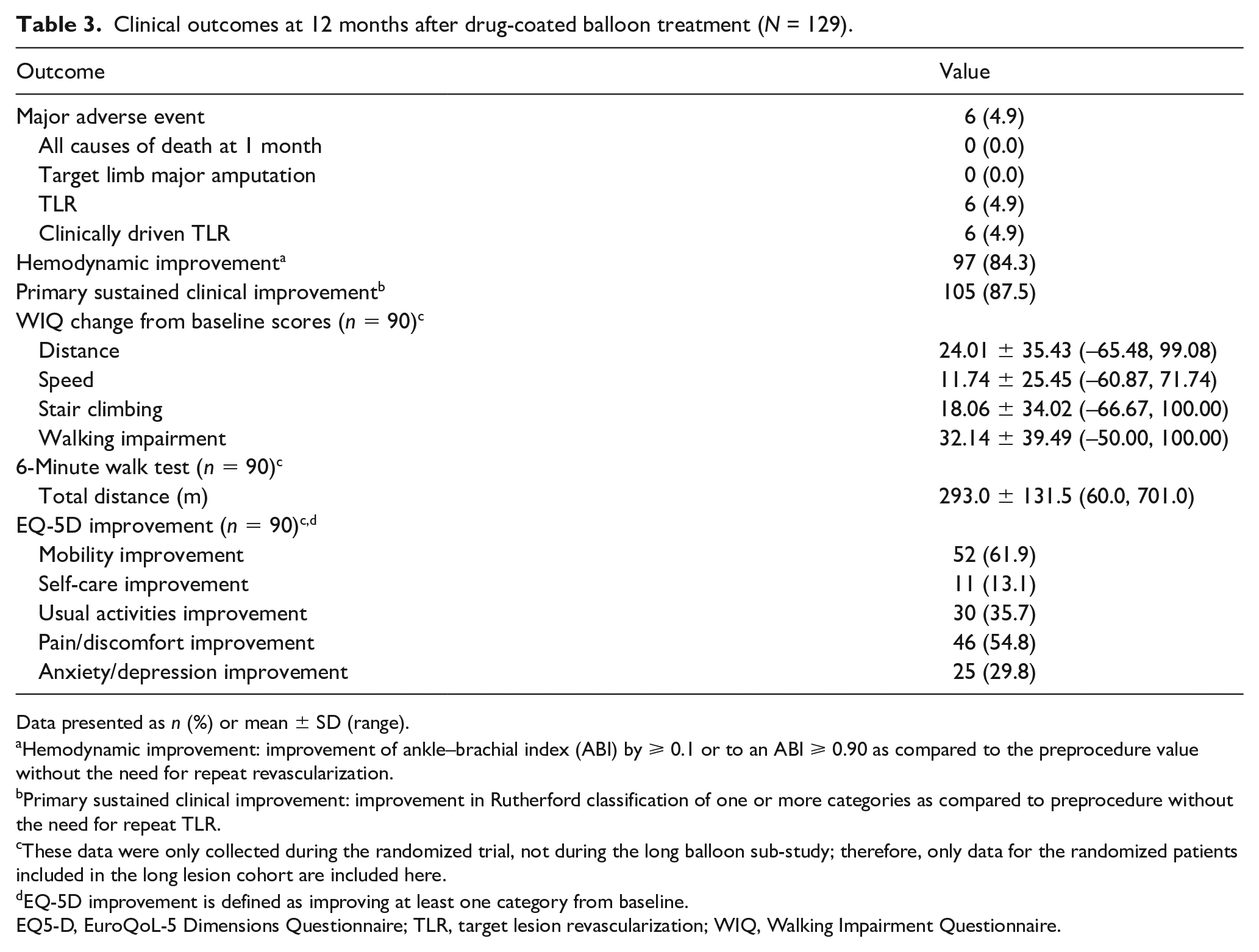
Data presented as n (%) or mean ± SD (range).
Hemodynamic improvement: improvement of ankle–brachial index (ABI) by ⩾ 0.1 or to an ABI ⩾ 0.90 as compared to the preprocedure value without the need for repeat revascularization.
Primary sustained clinical improvement: improvement in Rutherford classification of one or more categories as compared to preprocedure without the need for repeat TLR.
These data were only collected during the randomized trial, not during the long balloon sub-study; therefore, only data for the randomized patients included in the long lesion cohort are included here.
EQ-5D improvement is defined as improving at least one category from baseline.
EQ5-D, EuroQoL-5 Dimensions Questionnaire; TLR, target lesion revascularization; WIQ, Walking Impairment Questionnaire.
Secondary effectiveness, safety, and clinical outcome assessments
All MAEs in the long lesion cohort were CD-TLR (Table 3). The Kaplan–Meier estimate for freedom from TLR was 95.1% at 12 months (95% CI: 89.7%, 98.2%) (Figure 2B).
Of those with available Rutherford categorization at baseline and at 12 months, 87.5% (105/120) had improved by at least one category without reintervention; that is, they had primary sustained clinical improvement. At 12 months, 66.7% (80/120) and 17.5% (21/120) were Rutherford category 0 or 1, respectively. Distribution of Rutherford categorization over time is shown in Figure 3. Of 115 for whom ABI data were available, 84.3% (97/115) experienced hemodynamic improvement (Table 3).
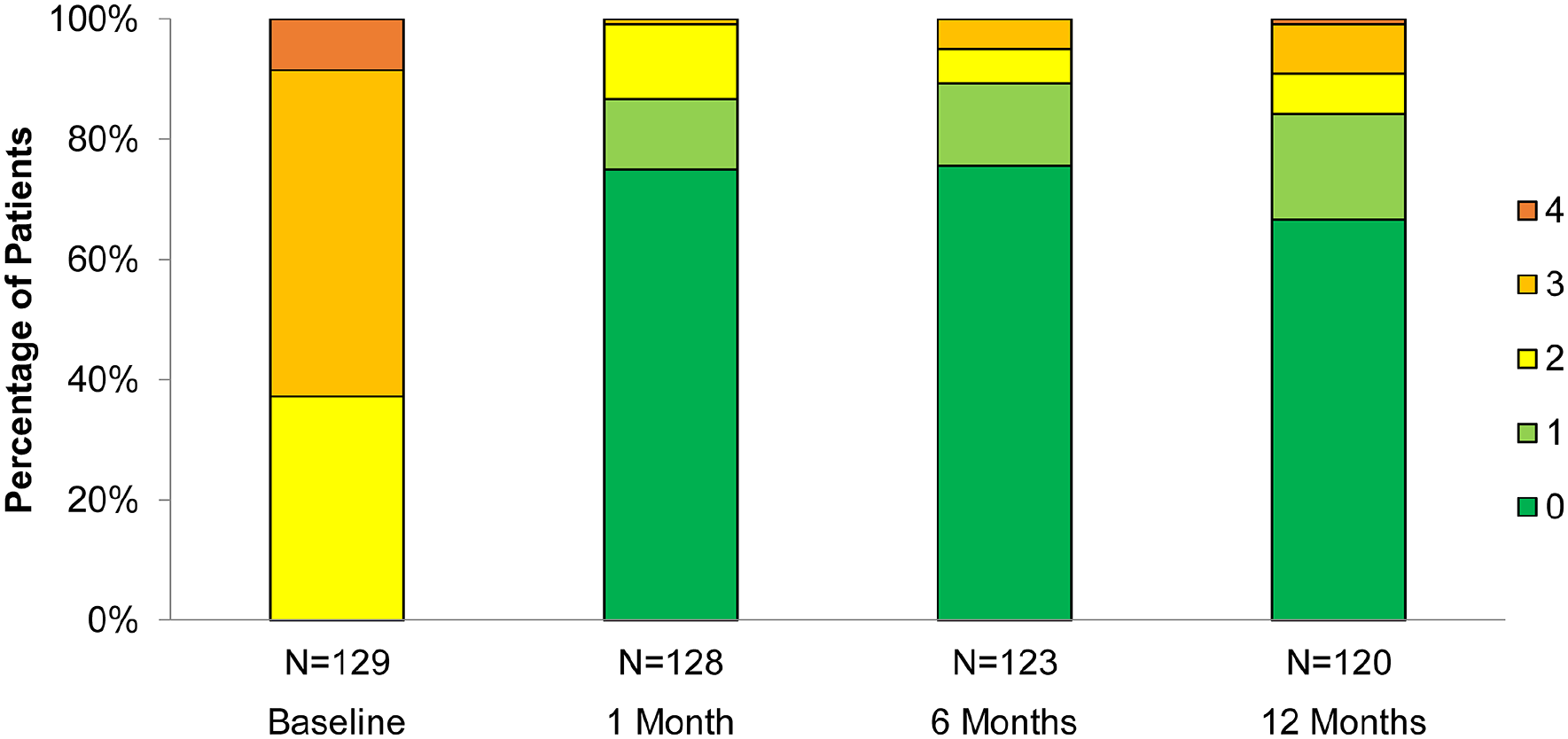
Rutherford category over 12 months post-procedure in the long lesion cohort. Colors represent the Rutherford categories.
WIQ, 6-minute walk test, and EQ5D measures were collected from patients enrolled in the RCT (n = 90 in the long lesion cohort); these results are summarized in Table 3.
Medication assessment
At 12 months posttreatment, 82.9% (97/117; 95% CI: 74.8%, 89.2%) of patients were using acetylsalicylic acid monotherapy; dual antiplatelet therapy was reported by 52.1% (61/117; 95% CI: 42.7%, 61.5%) at 12 months.
Discussion
Patients included in the RANGER II SFA long lesion cohort had lesions ranging in length greater than 100 mm, with a mean length of 144.5 mm. Despite the long lesion lengths, bail-out stenting was employed at low frequency (7%), which may have been facilitated by the long (3 minutes) inflation duration prescribed for the DCB. Patients demonstrated high primary patency rates at 1 year as well as a high rate of sustained clinical improvement. MAE rates were low, and all MAEs were CD-TLRs.
The long lesion cohort patients had 1-year safety and efficacy results similar to those previously reported for the overall RANGER II SFA RCT (mean lesion length 82.5 mm). 13 The CD-TLR rate for the RCT was 5.6% (5/90) for the Ranger DCB arm; for the long lesion cohort, it was 4.9%. Primary patency by Kaplan–Meier analyses was 89.8% in the RCT for the Ranger DCB; for long lesions, it was 88.0%. These observations suggest consistent results among patients treated with the Ranger DCB, including those with longer lesions.
Prior reports of DCB use in lesions > 100 mm mainly represent ‘real-world’ cohorts, including the SFA-Long study and the Lutonix Global SFA Registry (Figure 4).5,6,8,9 These studies had longer mean lesion length (e.g., mean lesion lengths for SFA-Long and Lutonix long lesion groups were 251.7 mm and 212.3 mm, respectively) compared to patients included in the present analyses. Bail-out stent use was notable at 10.9% in the SFA-Long study and 35.7% in the Lutonix DCB Global SFA Registry long lesion subgroup.5,8 Twelve-month patency in these studies was 83.2% and 65.2%, respectively.
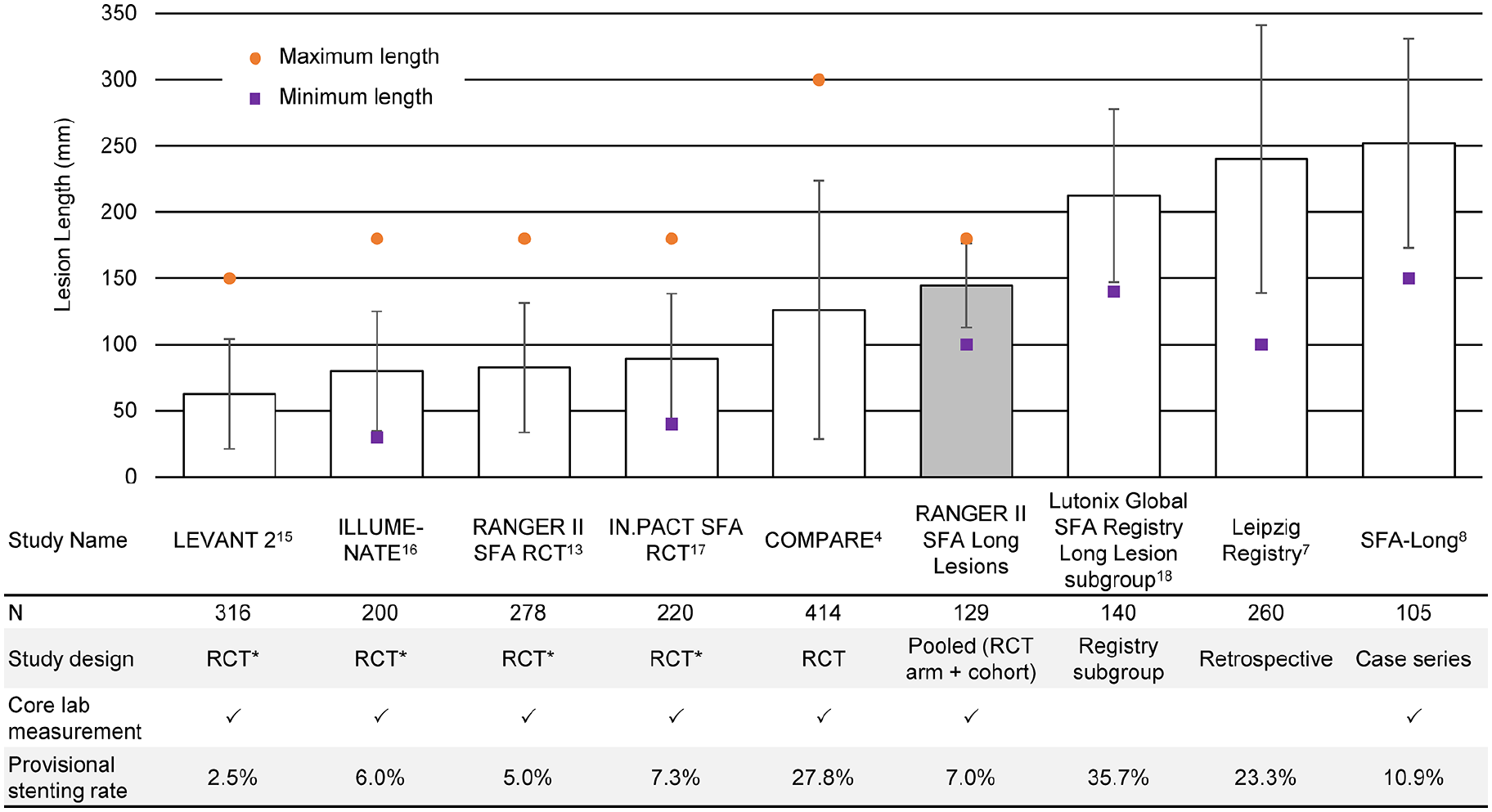
Lesion lengths in pivotal DCB trials13,15–17 and studies of ‘long’ lesions.4,7,8,18 Reported mean lesion length (SD) and minimum and maximum lengths per study eligibility criteria shown. The COMPARE study 4 stratified enrollment by lesion length and included 276 patients with lesions > 10 cm. Maximum lesion length for both the RANGER II SFA RCT and long balloon sub-study contributing to the long lesion cohort was 180 mm.
The COMPARE trial, which evaluated patients treated with the Ranger DCB or the higher-dose IN.PACT DCB (dose density of 3.5 µg/mm2; Medtronic, Minneapolis, MN, USA), also provides important context for long femoropopliteal lesions. 4 In this randomized noninferiority trial, overall results demonstrated no significant differences in safety with comparable excellent outcomes with both balloons. 4 Similar results between the low- and high-dose DCBs were also observed in patient strata with lesion length > 100 mm. 4 COMPARE mid-length stratum is similar to the lengths presented here, and both the mid- and long-strata showed similar patency to the RANGER II RCT and SFA-Long studies.9,13
Additional trials and real-world data are needed to validate the results from the RANGER II SFA long lesion cohort study. Although patients representing non-White racial and ethnic groups comprised nearly one-third of patients in this analysis—more than were reported in the aforementioned comparable studies—representation of these patient groups in clinical trials remains disproportionate. The ELEGANCE Registry (Clinical trials.gov identifier: NCT04674969) will contribute to building this body of evidence in real-world practice conditions. Replication of the analyses presented here using more complex real-world data would support the generalizability of these findings to other patient populations with long lesions.
Study limitations
Limitations of the RANGER II SFA long lesion cohort study include the fact that the study utilized a highly selected subset of patients from a clinical trial population, which may limit generalizability of the results to broader patient populations. The analysis did not include a comparator group. Adjunctive primary treatment modalities (e.g., laser, atherectomy, scoring/cutting balloons, and other debulking devices) were excluded from use during the index procedure. Additionally, the study was a post hoc secondary analysis of these data, with most long lesion patients having been treated with multiple overlapping balloons rather than one long balloon. The study was not sufficiently powered to analyze differences in patient outcomes associated with different balloon diameters, nor was it powered to explore differences between patients who were treated with one or multiple balloons. Such comparisons in future studies would provide information on the economy of using multiple balloons for long lesions as well as the clinical outcomes. The long lesions represented in the cohort were distributed across the SFA, but only one included the proximal popliteal segment; thus, further investigation is needed to confirm whether results translate to the frequently encountered real-world situation of long lesions in the proximal popliteal artery anatomy. Previously stented lesions were excluded, limiting translatability in cases of in-stent restenosis. Finally, the data used in these analyses included a short duration of follow-up as patients from the LBSS were only followed for 1 year; longer follow-up is needed to determine whether patency remains long-term in this patient population.
Conclusions
In conclusion, patients with lesions ⩾ 100 mm in length (mean 144.5 mm) treated with Ranger DCBs in the RANGER II SFA RCT and LBSS had 1-year safety and efficacy results similar to the previously reported results for the overall RANGER II SFA RCT cohort (mean lesion length 82.5 mm). These findings suggest that the Ranger DCB can provide consistent results regardless of lesion length.
Footnotes
Acknowledgements
The authors thank the following Boston Scientific employees (Maple Grove, MN, USA) for their assistance: Chelsea Liu, PhD (statistical analysis management), Phil Ghizoni, MBA (clinical trial management), and Elizabeth J Davis, PhD and Alexandra J Greenberg-Worisek, PhD, MPH (medical writing).
Data availability statement
Declaration of conflicting interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Schroë is a consultant for Boston Scientific. Dr Sachar is a consultant for Boston Scientific and Medtronic; in the last 24 months he has received research funding from Boston Scientific, Medtronic, Terumo, and Gore; he is a shareholder of Contego Medical. Dr Keirse has no relevant disclosures. Dr Soga has no relevant disclosures. Dr Brodmann is a consultant for BD Bard, Philips, Medtronic, Boston Scientific, Shockwave, Cook Medical, Cagent Vascular, Bayer Healthcare, Biotronik, Reflow Medical. Dr Rao has no relevant disclosures. Dr Werner has no relevant disclosures. Dr Holden is a Clinical Investigator and Medical Advisory Board Member for Boston Scientific. Dr Lopez has no relevant disclosures. Dr Krishnan has no relevant disclosures. Dr Diaz-Cartelle is a Medical Director at Boston Scientific.
Funding
RANGER II SFA was funded by Boston Scientific Corporation, Marlborough, MA, USA. Boston Scientific employees were involved with study management, data analysis planning, and manuscript preparation.