
Abstract
Aortic aneurysms were the primary cause of nearly 10,000 deaths in 2014 according to data from the Centers for Disease Control and may involve segments of the thoracic or abdominal aorta. Thoracic aortic aneurysms and dissections are more commonly associated with an underlying genetic etiology. In the past several decades, in parallel with the burst of new genome sequencing technologies, a number of genetic aortopathies have been identified. These have provided important insights into the molecular mechanisms of aneurysmal disease, but pose challenges in clinical practice as there are limited consensus recommendations at this time. In this review, we aim to address the pathophysiology, clinical presentation, and treatment considerations in the key heritable thoracic aortopathies.
Keywords
CME Opportunity
Continuing medical education (CME) credits and maintenance of certification (MOC) points are available for reading this article. This opportunity is made possible through a joint partnership with University of Virginia School of Medicine (UVASOM). For instructions, please visit the Society for Vascular Medicine’s website at
Accreditation Statement
The University of Virginia School of Medicine (UVASOM) is accredited by the ACCME to provide continuing medical education for physicians. UVASOM designates this journal-based CME activity for a maximum of one AMA PRA Category 1 Credit™. Participants should claim only the credit commensurate with the extent of their participation in the activity. UVASOM, as an accredited provider, awards 1 hour of participation (consistent with the one AMA PRA Category 1 Credit™) to participants who successfully complete this educational activity. Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to one MOC II point in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. It is the CME provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC II credit. UVASOM maintains a record of participation for 6 years.
Disclosures
The faculty, staff and planning committee of the University of Virginia Office of Continuing Medical Education have no financial affiliations to disclose. The CME planning committee disclosed the following: Heather Gornik is supported by CVR Global and receives intellectual property rights from Summit Doppler Systems, Inc. and intellectual property rights and stock/ownership from FlexLife Health. Aditya Sharma is supported by National Institute of Health Sciences, AstraZeneca, and Biomet Biologics. Geoff Barnes is supported by Pfizer/Bristol-Myers Squibb (BMS), Blue Cross Blue Shield of Michigan, and the National Heart, Lung, and Blood Institute and serves as a consultant for Pfizer/BMS, Janssen, and Portola. Valerie Clark has no financial affiliations to disclose. The authors have disclosed the following: Dr Kanthi has served as a consultant to Acer Therapeutics. The other authors have disclosed no financial relationship or interest with any proprietary entity producing healthcare goods or services.
Introduction
Aortic aneurysms are permanent, localized dilations of the aorta that exceed the normal aortic diameter by at least 50%. The most feared complications of aneurysms are rupture or dissection, which can be catastrophic. In a multinational study, aortic aneurysms accounted for greater than 160,000 deaths and 2.9 million disability-adjusted life-years globally in 2015.1,2 Recent studies have suggested the number of cases of aortic disease is increasing over time, which may reflect improved diagnostic modalities and/or increased awareness of aortic disease.3,4
Aortic aneurysms are broadly classified by their anatomic involvement of the thoracic (TAA) or abdominal aorta (AAA). AAAs are the most commonly identified aortic aneurysm, with the vast majority related to atherosclerotic risk factors. There is evidence of familial clustering in AAA and ongoing efforts to clarify the genetic basis for these findings, but currently these studies do not have clinical relevance. In contrast, TAAs are more likely to be associated with a genetic cause and are further subdivided into those involving the ascending aorta (60%), aortic arch (10%), descending aorta (40%) and thoracoabdominal aorta (10%). 5
The majority of genes associated with the development of TAA encode proteins involved in the extracellular matrix (ECM), vascular smooth muscle cell (VSMC) contraction or metabolism, and/or are involved in the transforming growth factor-beta (TGF-β) signaling pathway. Prior to recent advances in genomics, aneurysm formation and growth were attributed to structural weakness of the aortic wall resulting from dysfunctional ECM proteins, as in Marfan syndrome (MFS) and vascular Ehlers–Danlos syndrome (vEDS).6,7 However, recent studies suggest that a common pathway involving TGF-β may underlie the development of many aortic aneurysms and dissections. TGF-β is a regulatory cytokine produced by many cells, including those of the vessel wall. There is a growing body of evidence suggesting alterations in TGF-β signaling are a primary etiologic driver of many TAAs.8,9
TAAs are commonly stratified into either clusters of ‘syndromic’ disorders with extravascular organ involvement, or seemingly isolated aberrations denoted ‘non-syndromic’ disorders. 10 Table 1 briefly outlines these disorders based on the signaling pathway involved, genetic lesion, and characteristic clinical manifestations.
Syndromic and non-syndromic TAAs.
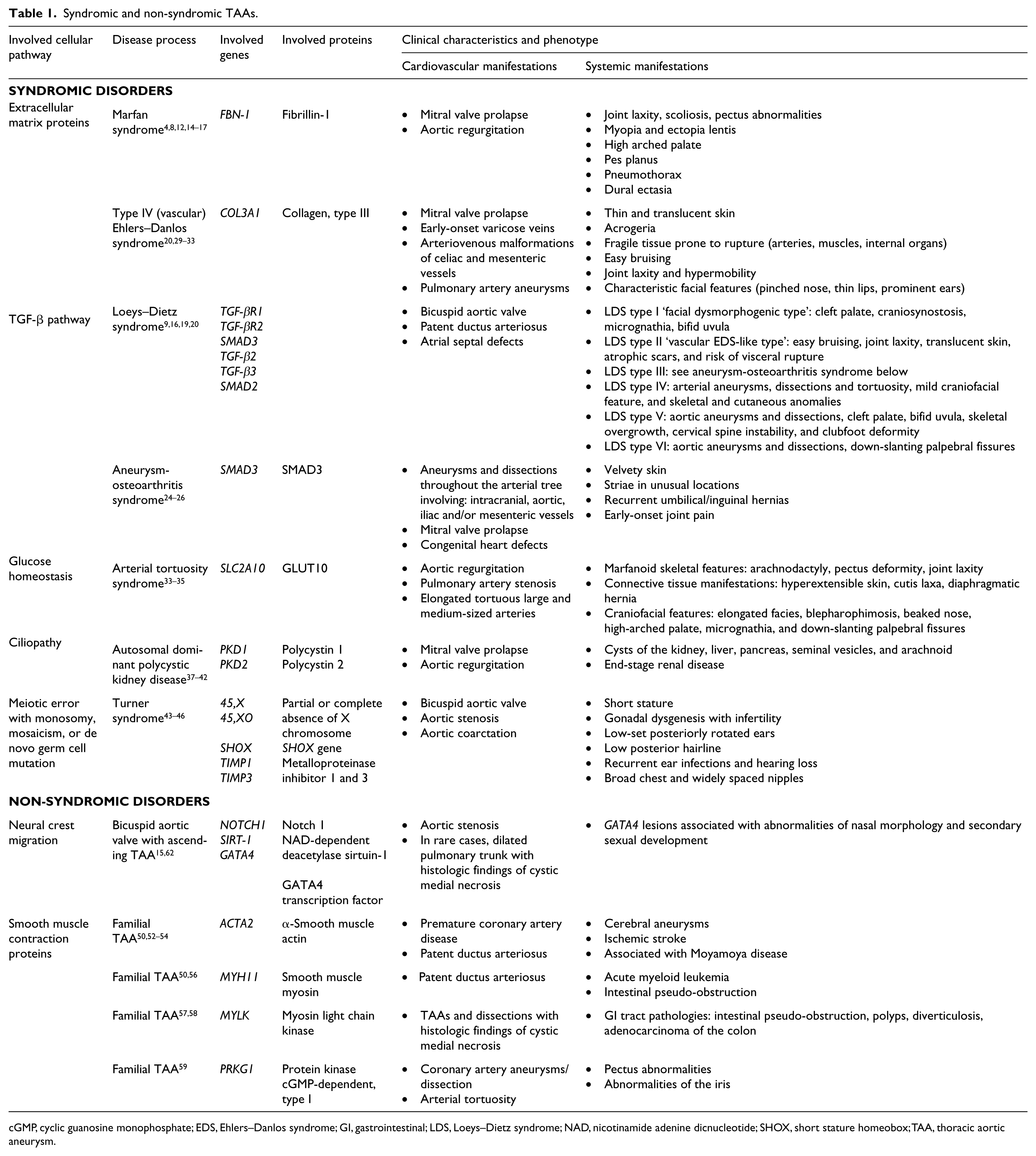
cGMP, cyclic guanosine monophosphate; EDS, Ehlers–Danlos syndrome; GI, gastrointestinal; LDS, Loeys–Dietz syndrome; NAD, nicotinamide adenine dicnucleotide; SHOX, short stature homeobox; TAA, thoracic aortic aneurysm.
Syndromic TAA
Patients with syndromic TAA often have characteristic features on physical exam that may help inform clinicians to assess for aneurysmal disease. The most well-described syndromic TAAs were detailed more than a decade ago. 11 These include MFS, Loeys–Dietz syndrome (LDS), aneurysm-osteoarthritis syndrome (AOS), vEDS, arterial tortuosity syndrome (ATS), autosomal dominant polycystic kidney disease (ADPKD) and Turner syndrome (TS). Despite the phenotypic overlap, these syndromes have different prognoses. Divergent median survival in patients with LDS (37 years), vEDS (48 years), and treated MFS (70 years) underline the importance of accurate diagnosis and optimized management strategies.9,12,13
Marfan syndrome
MFS is the best known aortic aneurysm syndrome and its cardiovascular features were first described systematically in 1955. 14 It is an autosomal dominant syndromic disorder affecting approximately one in every 5000 persons and implicated in 3–5% of all aortic dissections.4,15 MFS results from mutations in FBN-1, producing defects in the skeletal, ocular, and cardiovascular systems. Fibrillin-1 is a large extracellular matrix protein encoded by FBN-1, which assembles to form ECM microfibrils critical to maintaining connective tissue structural integrity.8,16 In addition to its structural role, fibrillin is essential in regulating cell signaling by sequestering TGF-β in the ECM. 8 The loss of fibrillin leads to increased bioavailable TGF-β and activation of both canonical SMAD-dependent and non-canonical SMAD-independent TGF-β signaling pathways, which then predisposes to aneurysmal dilation.8,16 The normal tissue distribution of fibrillin is reflected in the characteristic phenotype of ectopic lentis, joint hypermobility, arachnodactyly and aortic root dilation.
Aortic dilation in MFS typically occurs at the sinuses of Valsalva and the tubular portion of the ascending aorta to form a ‘pear-shaped’ annuloaortic ectasia. The primary cause of death in patients with MFS is progressive aortic root enlargement with subsequent dissection. The natural history of disease is underscored by the fact that 90% of untreated patients with MFS will develop an aortopathy that requires aortic surgery or will suffer a dissection. 17 Evidence of genotype-phenotype correlation is emerging that individuals with a truncating mutation in FBN-1 have a higher likelihood of suffering an aortic event at an earlier age than those with missense mutations. 18 Advances in medical management, including the use of beta-blockers and refined surgical techniques, have improved survival. 12
Loeys–Dietz syndrome
LDS is an autosomal dominant aortopathy characterized by aggressive TAAs. Several variants of LDS exist including types I–VI that vary according to which member of the TGF-β signaling pathway is dysregulated. These LDS variants and associated phenotypes are listed in Table 1. Patients with LDS may have aberrant craniofacial, skeletal, and cardiovascular features. 16 The original LDS diagnostic schema was based upon the severity of craniofacial versus cutaneous features; however, this was revised in 2014 and now defines LDS variants according to their underlying genetic lesion.19,20 There are data suggesting those with more pronounced craniofacial features which may be assessed by a craniofacial severity index score had cardiovascular complications at younger ages than those with less prominent features. 9
LDS type I, or Facial Dysmorphogenic LDS, is marked by prominent craniofacial features including cleft palate, craniosynostosis, micrognathia and/or bifid uvula. 20 LDS type II, or vascular EDS-like LDS, has less prominent craniofacial features but patients often have easy bruising, joint laxity, thin translucent skin, atrophic scars, and visceral rupture events. 20 LDS type III, also known as aneurysm-osteoarthritis syndrome, is discussed below. LDS types IV, V, and VI are less common and typically have a less severe clinical course. 20 Signaling by TGF-β is crucial to the development and maintenance of many tissues, including blood vessels and craniofacial growth and patterning. 16 Loss-of-function mutations in TGF-β receptors 1 and 2, SMAD-dependent intracellular signaling proteins, and/or TGF-β2 or 3 cytokines leads to increased overall TGF-β activity and predisposition to the vascular, craniofacial, and skeletal manifestations seen in LDS.16,21
The vascular mortality of LDS is dramatic and TAA in LDS can grow at rates greater than 1.0 cm/year, nearly 10-fold higher than the average growth of aneurysms in MFS. 22 In one series of patients with LDS, the mean age of death was 26 years old, with TAA dissection implicated in 67% of deaths, AAA dissections in 22%, and intracranial bleeding events in 7%. 9 Thus, for most patients with LDS early prophylactic surgery is recommended. 23
Aneurysm-osteoarthritis syndrome
AOS is an autosomal dominant mutation in SMAD3, which encodes an intracellular effector of the TGF-β signaling pathway. 24 The clinical phenotype of AOS includes overlapping craniofacial and skeletal features of MFS and LDS, early-onset joint abnormalities, arterial tortuosity and aneurysms and predilection for aortic dissection. 25 In one study, the most common presenting symptom for patients with AOS was early-onset joint pain and of the patients initially presenting with joint pain 20% died suddenly from aortic dissections. Further, nearly 90% were found to have arterial abnormalities diffusely in the vascular tree, including in intracranial, aortic, and intra-abdominal arteries.25,26 There are no current consensus guidelines on management, but given the aggressive behavior of AOS, early elective aneurysm repair has been advocated. 26
Vascular Ehlers–Danlos syndrome
Vascular EDS, or EDS type IV, is an autosomal dominant disorder of COL3A1, which encodes type III collagen, an important architectural component of the connective tissue in skin, blood vessel walls, and visceral organs. These patients consequently may have thin translucent skin, fragile soft tissues prone to rupture, and joint laxity. Further, degradation of collagen leads to loss of tensile strength in arteries, resulting in vascular fragility. The prevalence is estimated to be between one in 10,000 to 25,000 in the United States, with most going undiagnosed until they develop a vascular complication. 27
Vascular EDS is one of 13 EDS variants in the updated 2017 EDS nosology and accounts for less than 5% of all EDS diagnoses, but carries the worst prognosis of all subtypes. 28 The diagnosis of vEDS can be challenging as patient selection for genetic testing relies on diagnostic criteria that has not been rigorously tested. 29 The Villefranche criteria is often employed and a recent retrospective review found it had 92% sensitivity but only 60% specificity in detecting symptomatic probands. 29 However, there is some evidence of genotype-phenotype correlation with vEDS severity and age of onset. Mutations that result in minimal (10–15%) normal type III procollagen production (usually missense mutations or exon splicing errors) had an earlier age of disease onset with a higher incidence of visceral artery pathology. 30 Whereas, mutations that lead to protein-truncation (nonsense or frameshift mutations) with half the amount of normal type III procollagen result in later age of disease onset but higher incidence of aortic pathology. 30 Therefore, genotyping in vEDS could directly impact approaches on clinical management and family screening.
One in four patients diagnosed with vEDS develop a significant health problem by age 20 and more than 80% develop life-threatening complications by age 40. 31 Vascular EDS has a surgical mortality of approximately 40% largely attributed to tissue fragility, poor wound healing, and perioperative bleeding complications. 32 As a result, surgical intervention should be planned and performed under controlled environments to improve safety.
Arterial tortuosity syndrome
ATS is a rare autosomal recessive disorder caused by loss-of-function mutations in SLC2A10, which encodes the facilitative glucose transporter GLUT10 important in glucose homeostasis. 33 The disease is characterized by arterial tortuosity, elongation, and stenosis of large and medium-sized arteries with a propensity for aneurysm formation, dissection, and ischemic events. 33 Affected patients typically have marfanoid skeletal features, connective tissue manifestations, and craniofacial features (see Table 1).
The prognosis of patients with ATS is poor, with mortality rates as high as 40% in the first 5 years of life. 34 However, a less aggressive phenotype with fewer vascular complications has recently been identified. 35 As the disease is rare and often leads to early childhood death there is a dearth of consensus clinical guidance. We direct readers to an excellent review for a more detailed discussion of ATS. 36
Autosomal dominant polycystic kidney disease
ADPKD is an inherited condition defined by the pathologic development of fluid-filled cysts in the kidney and subsequent chronic kidney disease. 37 ADPKD is the most commonly inherited kidney disease affecting approximately one in 1000 individuals. 38 In addition to renal cystic disease, patients with ADPKD may have hepatic cysts, valvular heart disease, and cerebral or thoracic aneurysms. In nearly 90% of cases, ADPKD is caused by mutations in PKD1, which encodes for the protein polycystin-1.
There is a fivefold increased risk of aortic aneurysm and dissection in patients with ADPKD compared to unaffected patients. 39 The similarities in body frame (tall and slender with lower total body weight) amongst patients with ADPKD and MFS has raised the possibility of a potential relationship between these diseases.40,41 Further, TAA in MFS and ADPKD most commonly involve the aortic root at the level of the sinus of Valsalva, whereas this is an unusual finding in TAAs associated with hypertension. 42
Turner syndrome
TS is a genetic condition affecting women caused by partial or complete monosomy of the X chromosome. TS is a relatively common chromosomal disorder, affecting approximately 1 in 2500 live female births. Patients affected have short stature, early-onset ovarian failure, metabolic and hormonal aberrations often leading to obesity and congenital cardiac abnormalities including bicuspid aortic valve (BAV) and aortic disease. 43 The molecular mechanisms of TS are still unclear; however, TS has been associated with haploinsufficiency of the SHOX gene on X-chromosomes that is essential in early embryologic development. 44 In addition, loss of TIMP1 and TIMP3 on the X-chromosome has been associated with concomitant BAV and TAA in patients with TS. 45 The incidence of aortic dissection in TS patients is 100-fold greater than unaffected women of similar ages and typically occurs in the third or fourth decade of life. 46 Because women with TS are often of short stature, determination of appropriate aortic size for prophylactic surgery has been challenging and many patients will suffer an aortic dissection with aneurysms measuring < 5 cm in diameter. 46 In these patients, the use of aortic size indexed by body surface area (BSA) or aortic size index (ASI) may offer a more accurate means of screening patients who require surgery. 47 For further discussion and recommendations on the cardiovascular management of TS, we recommend an excellent AHA Scientific Statement from 2018. 48
Non-syndromic TAA
In recent years, there has been a growing recognition that a subset of patients exhibit TAAs without evidence of overt connective tissue disorders or bicuspid aortic valve. These non-syndromic aneurysms can be clustered as familial TAA (FTAA) or sporadic TAA (sTAA) based on the presence of affected family members. FTAA typically present earlier in life than sporadic aneurysms, have a higher annual growth rate, and are not associated with traditional risk factors for aortic disease.49,50 Thoracic aortic aneurysms and dissections (TAAD) are frequently familial diseases and recent studies have noted that among patients with a known TAA and without evidence of a vascular connective tissue disorder, there is an approximately 20% incidence of arterial aneurysm in at least one first-degree family member. 51
Familial TAA
Patients with confirmed TAA and no known history of collagen vascular disorders or BAV, frequently have family pedigrees with multiple family members affected by TAA. 49 Modern genome sequencing technologies have identified a number of genetic lesions associated with the development of TAA that show familial aggregation. These FTAAs are a genetically heterogeneous population of disorders usually with autosomal dominant inheritance patterns and commonly involving defects in vascular smooth muscle function.
VSMCs are parallel layers of elastic fibers that form concentric layers of lamellar units in the media with a contractile apparatus composed of thick and thin filaments. This contractile apparatus allows blood vessels to alter vascular tone. The most abundant protein in VSMCs is smooth muscle α-actin encoded by ACTA2. The myosin superfamily is composed of large motor molecules, which are hexameric complexes of myosin heavy and light chains that form the ‘thick filaments’ that interact with actin ‘thin filaments’ to result in smooth muscle contraction. The myosin heavy chains in these complexes are encoded by MYH11. Myosin light-chain kinase is encoded by MYLK, an enzyme which enables normal smooth muscle myosin interaction with actin filaments for contraction. Finally, smooth muscle relaxation occurs when nitric oxide excreted by the endothelium induces VSMC cyclic guanosine monophosphate (cGMP)-dependent protein kinase to dephosphorylate myosin light-chain kinase and produces smooth muscle relaxation. Dysfunction of the VSMC contractile apparatus at these key steps impairs resilience of aorta in response to pulsatile blood flow or shear stress forces and consequently represents a few of the known FTAAs.
ACTA2
Autosomal dominant loss-of-function mutation in ACTA2, which encodes a specific smooth muscle α-actin isoform involved in VSMC, is the most common genetic cause of TAA and accounts for 10–15% of all FTAA. 52 These mutations interfere with the ability of arteries to stretch, resulting in FTAA. 53 However, reduced penetrance and variable disease expression results in half of mutation carriers having no aortic disease. 50 ACTA2 mutations may be associated with thoracic aortic and neurovascular aneurysms, patent ductus arteriosis (PDA), and other complications in smooth muscle-dependent organs. 54 In particular, missense mutations in ACTA2 that disrupt Arg258 have been associated with aggressive cerebrovascular disease with case reports of dilation of the proximal internal carotid arteries, occlusive disease of terminal internal carotid arteries, and abnormally straight course of intracranial arteries that predispose to cerebral ischemia. 55 The majority of ACTA2 aortic dissections occur in aneurysms less than 5 cm in size. Early surgical intervention is often considered in patients with ACTA2 mutations with even minimal change in aortic diameter. 52
MYH11
MYH11 encodes myosin heavy chain 11 and defects in this gene are associated with FTAA, particularly ascending TAA in association with PDA. These mutations are thought to account for 2% of non-syndromic TAA.50,56
MYLK
MYLK encodes myosin light-chain kinase and is associated with a familial syndrome characterized by acute aortic dissection, often with absent or very small preceding aneurysms. 57 In one case series of an affected family, all those affected presented with acute aortic dissections at variable ages with little to no enlargement of the aorta. 58
PRKG1
PRKG1 encodes type I cGMP-dependent protein kinase, which is responsible for smooth muscle cell relaxation. Mutations in this gene are associated with coronary aneurysms and aortic dissections that often present at a young age. In one study, aortic dissections were discovered in patients as young as 15 years old and occurred at aortic diameters as small as 4.3 cm. 59
Sporadic TAA
Sporadic TAAs are non-germline mutations and the exact mechanisms of disease remain poorly understood at this time. However, while specific familial mutations have not been identified in sTAAs, studies have reported up to 20% co-occurrence of TAA in first-degree family members, suggesting that there may be an as yet unidentified heritable component of disease. 49
Several of the sTAAs have been discovered while studying aortic tissues in patients affected by thoracic aneurysms or dissections. In one such study, a number of aberrantly regulated microRNAs were associated with the development of aortic disease. 60 In addition, regional tissue expression of matrix metalloproteinases (MMPs) responsible for normal tissue turnover of the ECM have been associated with TAAD. In particular, a mouse model of TAAD showed that increased levels of an MMP called ADAMTS-4 that is expressed in aortic tissue was associated with increased VSMC apoptosis, inflammatory cellular infiltration, and the development of sporadic TAAD. 61
Bicuspid aortic valve
BAV is the most common developmental cardiovascular malformation, affecting between 0.5% and 1.0% of the general population. Approximately 40–50% of patients with BAV also have aortic root and ascending aorta dilation. 15 Additionally, these patients may also have coarctation of the aorta or intracranial berry aneurysms. The exact mechanism of aneurysm formation is unclear and the etiology is likely polygenic with incomplete penetrance. 15 However, some cases have been associated with aberrant NOTCH-1 and SIRT-1 signaling or mutations in the GATA family of transcription factors. 62 Increases in proximal aortic shear wall stress forces with a bileaflet rather than trileaflet aortic valve also contribute to aneurysm formation and propagation.
Fibromuscular dysplasia
FMD is a non-inflammatory and non-atherosclerotic disorder that can lead to arterial stenosis, aneurysm, dissection and tortuosity. Approximately 80–90% of patients with FMD are women. 63 FMD most commonly affects the carotid and renal arteries, although any arterial bed may be involved. In a registry of patients with FMD, 2% of patients developed an aortic aneurysm, higher than would be expected in a predominantly young, non-smoking female population.64,65 The development of FMD is thought to be multifactorial with genetic and environmental influences.63,66 In a recent genome-wide association study, a single nucleotide polymorphism in the PHACTR1 locus conferred an odds ratio of 1.3 for FMD. 67 This locus is also associated with coronary artery disease, migraine, and cervical artery dissection. 67 Further studies are needed to clarify the genetic basis for FMD.
In addition to the disease processes described above, a number of other non-heritable disorders are implicated in aneurysm formation, including: tobacco use, renin-angiotensin-aldosterone system mediated aneurysms, vasculitides, atherosclerosis and infections of the aortic wall. Table 1 outlines some of the key heritable aortopathies.
An approach to diagnosis
In making the diagnosis of heritable aortopathies, an approach to history taking and physical exam, as well as knowledge of when to screen patients for these genetic disorders, is important. Heritable TAAs often occur in younger patients without apparent risk factors for atherosclerotic disease.
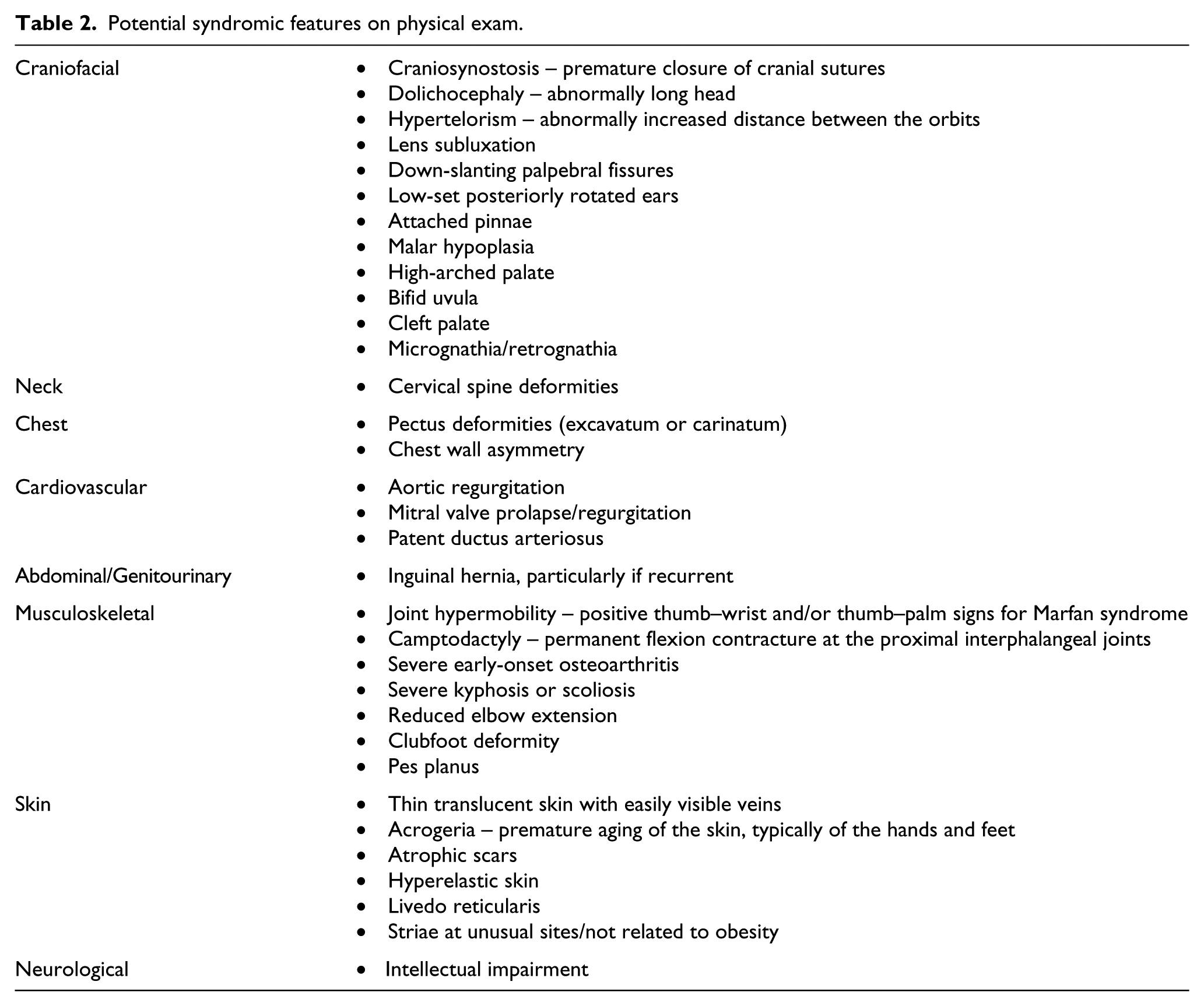
A detailed family history is paramount in making the diagnosis of heritable TAAs. The Modified Ghent criteria for MFS highlights this fact with separate criteria for the diagnosis of MFS in patients with family history compared to isolated non-familial cases. Because of reduced penetrance and expressivity of many of the heritable TAAs, it is recommended that detailed three-generation family histories be obtained for patients with TAA or dissection, aneurysms in any location of the arterial tree, left-sided congenital heart defects and sudden cardiac death before the age of 45. 68 Figure 1 shows an example of a pedigree of a patient with an FBN-1 mutation. Further, because there is significant phenotypic overlap among the syndromic TAAs, there are typical clinical features that should be routinely evaluated. Table 2 lists many of these potential syndromic features.
Potential syndromic features on physical exam.
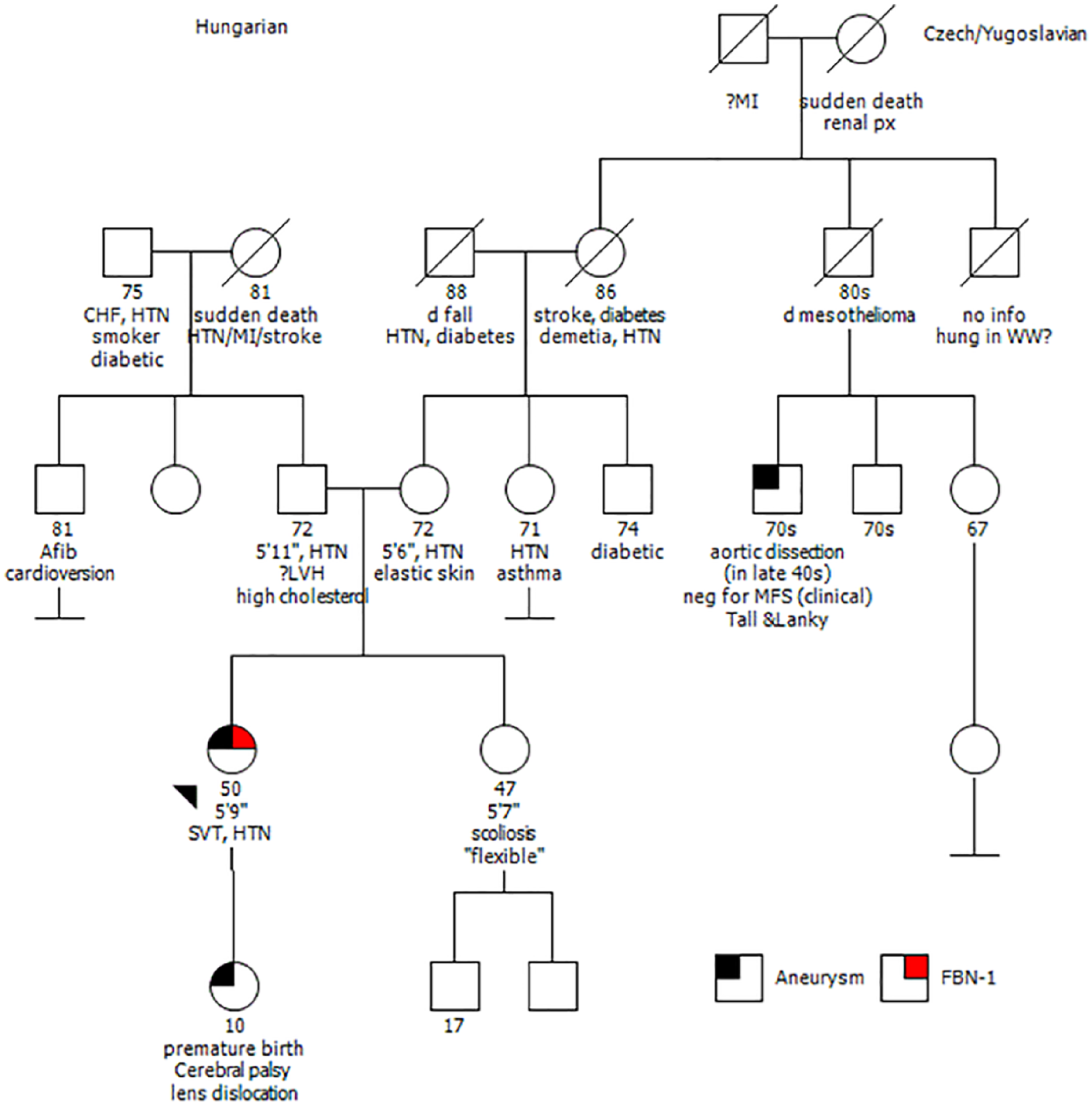
An example of a detailed 5-generation pedigree of a patient with an FBN-1 mutation with the proband marked by the black arrowhead. The squares represent males and circles represent females in this family. Diagonal lines indicate deceased members. The numbers below each shape indicates the known or approximated age in years at the time of pedigree analysis.
The clinical presentations of LDS and MFS can be similar, but there are important differences. Their common clinical features include: scoliosis, pes planus, chest wall deformities, pneumothorax, dural ectasia and aortic root dilation. 20 Figure 2 displays a CT scan of a characteristic pear-shaped aortic root aneurysm in a patient with MFS. There are a number of clinical features that are associated with LDS but not seen in patients with MFS, including: craniosynostosis, hypertelorism, abnormal uvula morphologies (bifid or broad), cleft palate, cervical spine instability, clubfoot, joint contractures and recurrent hernia. 20 Furthermore, aneurysms in MFS predominantly affect the aortic root and ascending aorta, whereas aneurysms in LDS may also involve the remainder of the aorta, aortic side branches, cerebral vessels or mesenteric arteries. 20 Additionally, there is no known association between LDS and ectopia lentis, which can be a key distinguishing feature between the two disease processes. 20
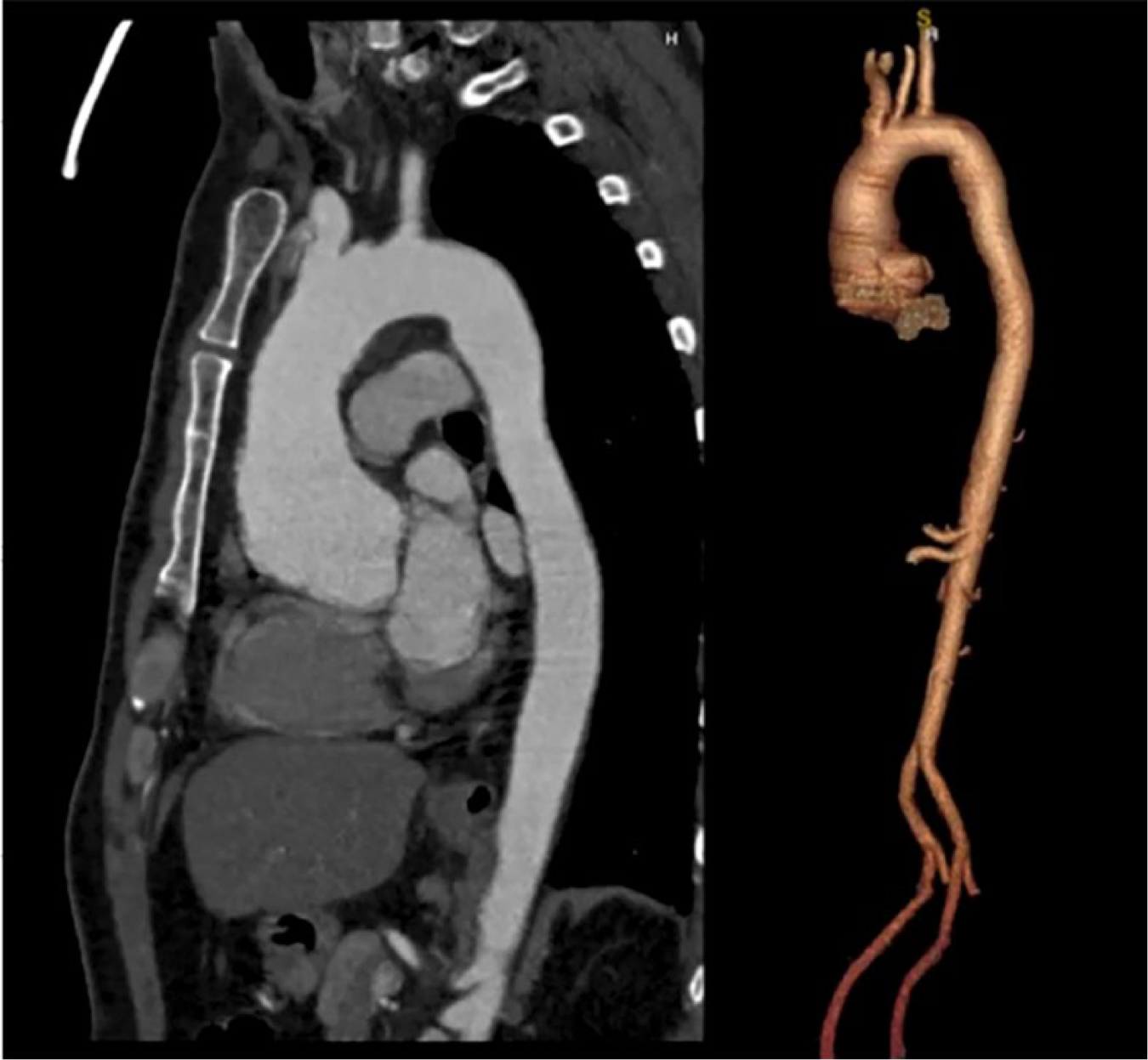
Sagittal (left) and 3D reconstruction (right) CT angiogram shows aortic aneurysm that measured 54 mm at the sinus of Valsalva in a patient with Marfan syndrome.
In patients with suspected MFS, the revised Ghent criteria should be employed as a high pre-test probability of disease confers 66–91% odds of finding an FBN-1 mutation and targeted FBN-1 testing should be pursued. 68 Tables 3 and 4 list the revised Ghent criteria. 69 However, if other familial TAAs are suspected, then the European Society of Human Genetics recommends testing for the following ‘core genes’: ACTA2, COL3A1, FBN-1, FLNA, MAT2A, MFAP5, MYH11, MYLK, NOTCH1, PRKG1, SMAD3, TGF-β2, TGF-β3, TGF-βR1 and TGF-βR2. 70 The ACCF/AHA has a Class I recommendation of screening first-degree family members of a patient with the following confirmed heritable TAAs: FBN-1, COL3A1, ACTA2, MYH11 and TGF-βR1. If there are no confirmed family members with syndromic TAAs, but a non-syndromic TAA is suspected, the ACCF/AHA have a Class IIa recommendation of screening for the ACTA2 gene. In practice, many institutions have bundled genetic panels, including many of these conditions that may be ordered, and are often less expensive than ordering each assay individually.
Revised Ghent criteria systemic scoring. 69
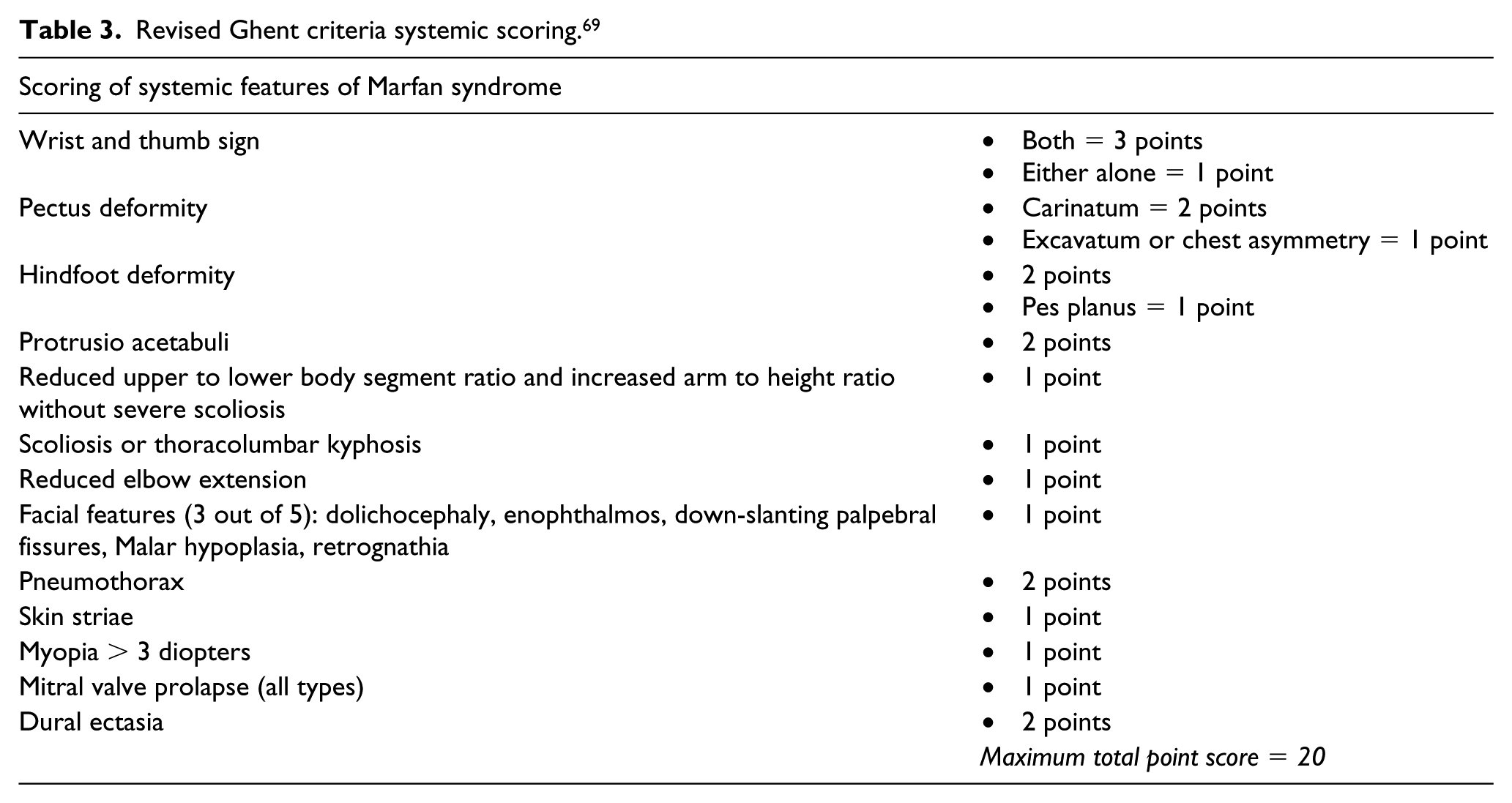
Revised Ghent diagnostic criteria. 69
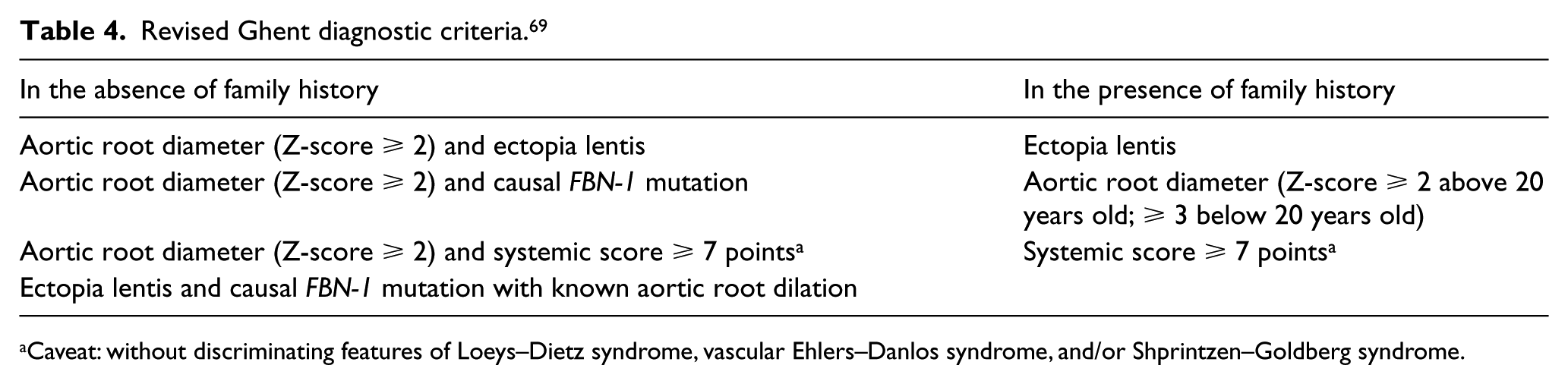
Caveat: without discriminating features of Loeys–Dietz syndrome, vascular Ehlers–Danlos syndrome, and/or Shprintzen–Goldberg syndrome.
There are no current consensus guidelines on TAA surveillance imaging frequency or preferred imaging modality. The European Society of Cardiology (ESC) thoracic aortic guidelines acknowledges that further investigation is required to guide this clinical decision. 70 We suggest obtaining surveillance imaging two to three times in the first 12–18 months of diagnosis to establish an aortic aneurysm growth velocity. Figure 3 shows this proposed algorithmic approach to surveillance imaging and management. In regard to choice of imaging modality, transthoracic echocardiogram (TTE), computed tomography (CT), and magnetic resonance angiography (MRA) are all considerations. TTE allows visualization of the aortic root and proximal ascending aorta, concomitant evaluation of potential associated cardiac valve disease, and will not expose the patient to radiation. However, TTE image quality is dependent upon sonographer expertise and patient body habitus. In contrast, aorta-protocoled CT imaging may allow for evaluation of the entire thoracic and abdominal aorta, but will expose the patient to radiation and dye contrast. Similarly, MRA with gadolinium contrast enhancement can be useful in visualizing the walls of the cardiac chambers and blood vessels, including coronary arteries. However, gadolinium can be retained in tissues with repeat imaging and lead to toxicity. 71 Alternatively, MRA with black blood gating may be employed to avoid exposure to gadolinium contrast.
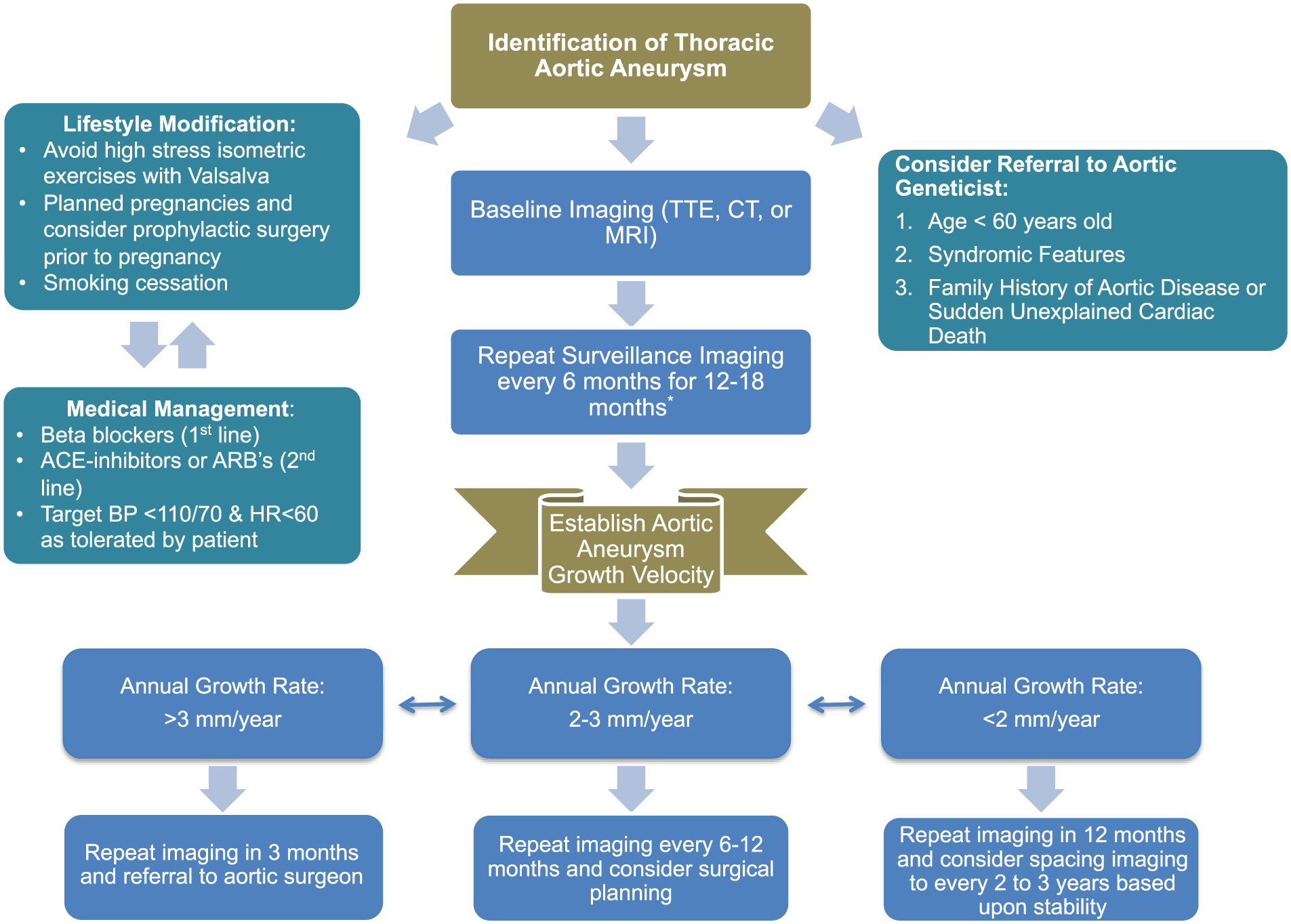
Proposed approach to management of TAA’s with attention to medical management, suggested lifestyle modifications, recommended criteria for referral to aortic geneticists and surveillance imaging frequency based upon aortic aneurysm growth velocities.
The importance of a multidisciplinary approach to diagnosis with input from medical and surgical aortopathy experts, qualified genetic counselors, and appropriate genetic testing cannot be understated. Genetics consultation can help address insurance coverage issues, guide family screening endeavors, and discuss reproductive planning. Further, as our ability to identify genetic variants currently outpaces our means to interpret these findings, genetic counselors can be invaluable resources to translate genetic information to clinical relevance in a beneficial, ethical, and sensitive manner for patients and their families. Not infrequently, genetic testing will result in ‘variants of uncertain significance’ (VUS) and genetics experts can provide recommendations on further testing that may be warranted to help clarify these results. Figure 4 illustrates many of these key issues that can be assessed in consultation with a genetics expert.
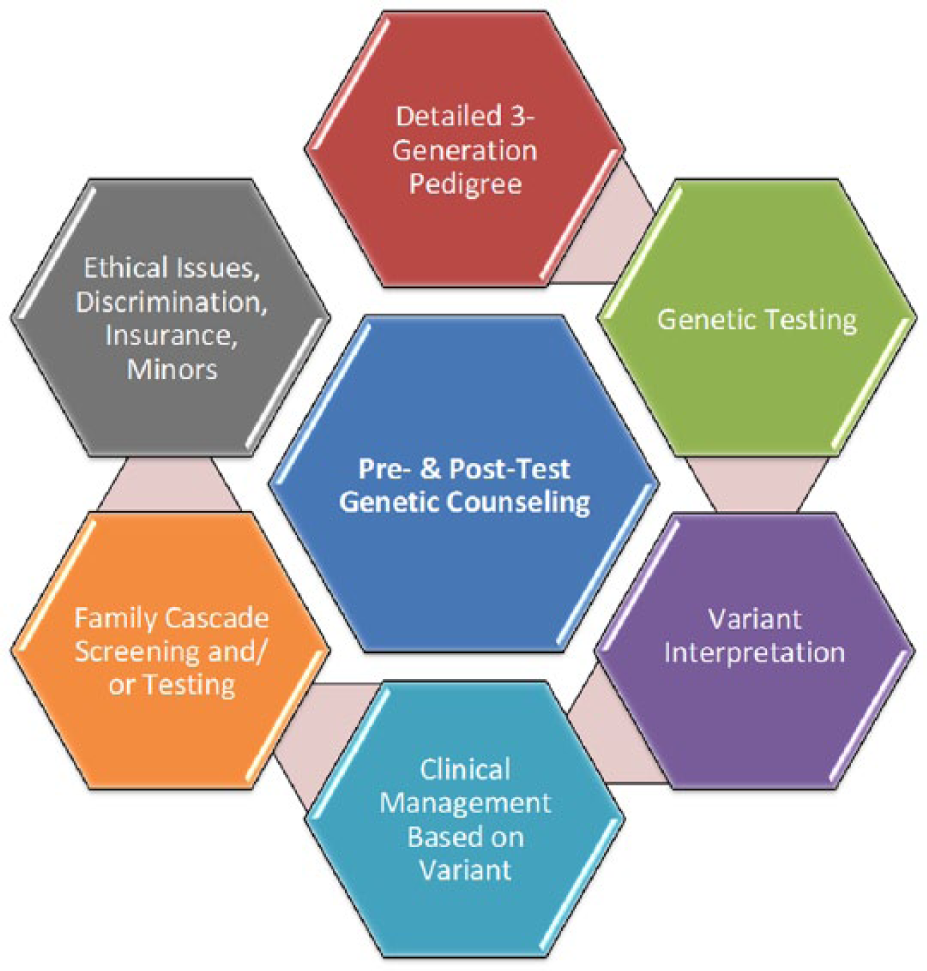
Genetic counselors and medical geneticists provide important insights into patient care prior to and following genetic testing.
Treatment and long-term management
In TAA of all sizes, the ACCF/AHA guidelines have a Class I recommendation of stringent blood pressure control with a goal of < 140/90 mmHg and smoking cessation. However, in clinical practice, many will target lower blood pressures, and sometimes as low as tolerated by the patient. In addition, vigorous isometric exercises that increase aortic wall stress increase the risk of aneurysm expansion and dissection. 23 In patients with aortic dissections or MFS, ‘anti-impulse therapy’, or reductions in both heart rate and systolic blood pressure with beta-blockers, has been shown to prevent dissection deaths in animal models and is considered a cornerstone of management. 72 This practice is generally employed for other aortic aneurysms as well. In our practice, we aim to maintain blood pressures < 110/70 mmHg and pulse rates < 60. Recent studies suggest that blocking the angiotensin receptor may have a role in aortopathy management, although further validation is needed. 73 Initial mouse models of MFS suggested that treatment with losartan blocked aneurysm formation; however, subsequent studies showed that losartan delayed but did not prevent deaths from aneurysm dissection. 74 Several clinical trials have shown there is no significant difference in aortic dilation with the use of beta-blockers compared with angiotensin receptor blockers in patients with MFS.73,75,76 There is preclinical evidence that drugs that lower blood pressure by decreasing smooth muscle contraction, such as calcium channel blockers or hydralazine, should not be used as first-line therapies.77,78 In aggregate, current evidence from multiple clinical trials supports the use of beta-blockers as first-line therapy, with angiotensin receptor blockers used in conjunction with beta-blockers, or as second-line therapy if beta-blockers cannot be tolerated. 79
The aim of surgical management of aortopathies is to intervene with prophylactic aortic repair prior to potentially devastating aortic dissection or rupture events. Table 5 lists the current surgical indications by disease for prophylactic intervention in TAAs.
Aggregated surgical indications for TAA repair in heritable aneurysmal diseases.
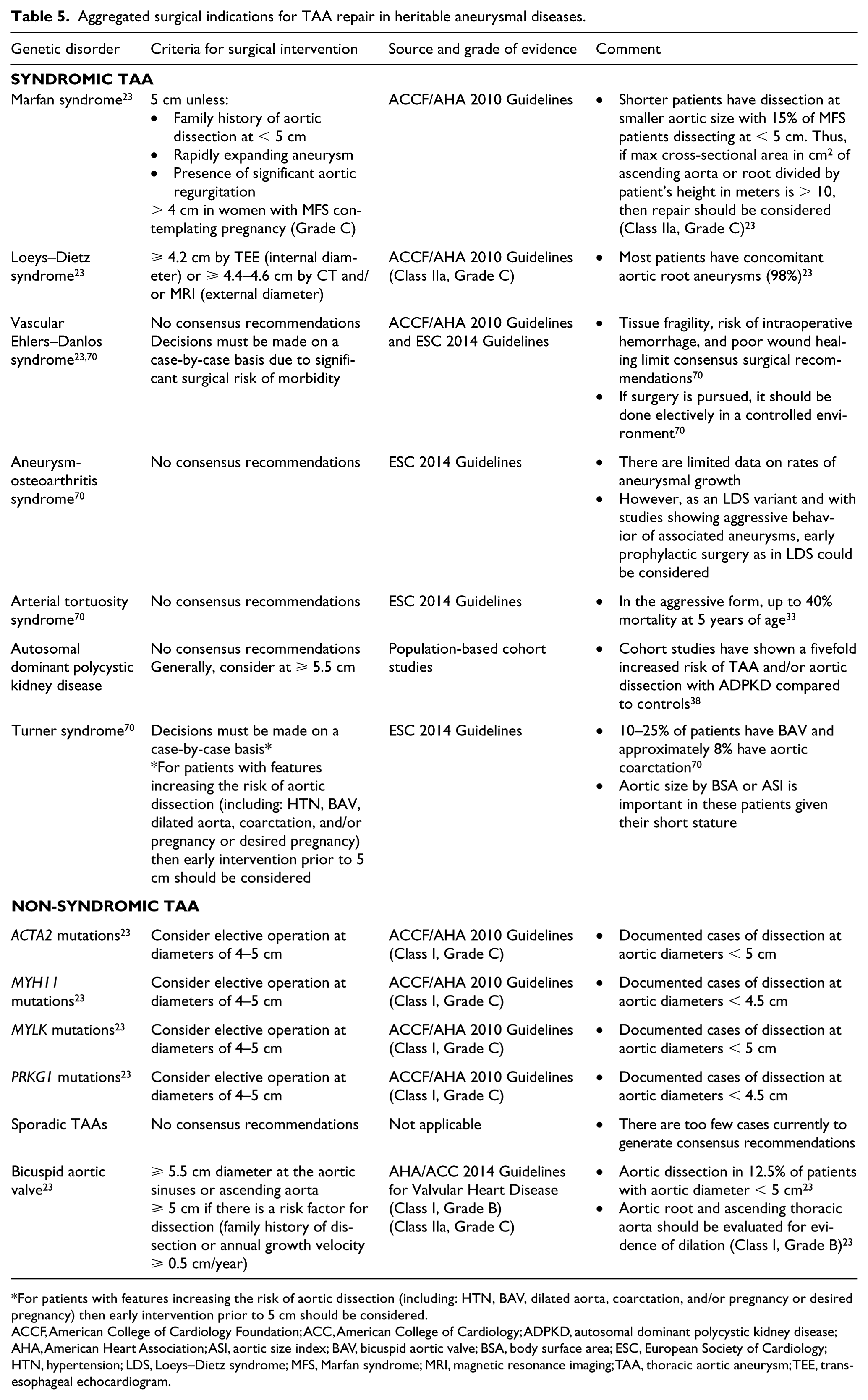
For patients with features increasing the risk of aortic dissection (including: HTN, BAV, dilated aorta, coarctation, and/or pregnancy or desired pregnancy) then early intervention prior to 5 cm should be considered.
ACCF, American College of Cardiology Foundation; ACC, American College of Cardiology; ADPKD, autosomal dominant polycystic kidney disease; AHA, American Heart Association; ASI, aortic size index; BAV, bicuspid aortic valve; BSA, body surface area; ESC, European Society of Cardiology; HTN, hypertension; LDS, Loeys–Dietz syndrome; MFS, Marfan syndrome; MRI, magnetic resonance imaging; TAA, thoracic aortic aneurysm; TEE, transesophageal echocardiogram.
Exercise restrictions
Exercise can lead to elevations in blood pressure that produce increased shear and pulsatile wall stress forces and cause aortic dissection or rupture in patients with aortic aneurysms. However, particular exercise restrictions for patients with aortic aneurysms have not clearly been delineated. Currently, most clinicians recommend avoiding high-stress isometric exercises, which can produce marked acute elevations in blood pressure. 80 In an analysis of 31 patients who suffered an acute aortic dissection during exercise, every case occurred with weightlifting or similarly strenuous exercise. 80 There is no evidence-based guidance on exercise restrictions, but most clinicians extrapolate findings from these weightlifting studies and recommend a 25–50 lb (approximately 10–25 kg) weight limit for patients.
Implications in pregnancy
There are often significant changes in cardiovascular hemodynamics during pregnancy, when maternal blood volume, heart rate, blood pressure, stroke volume, and cardiac output increase. 81 Further, pregnancy induces changes in plasma estrogen and progesterone levels that lead to changes in the aortic media, rendering the aorta more vulnerable to aortic dissection or rupture. Acute aortic events most commonly occur in the third trimester (50%), or in the peripartum period (33%). 82 The majority of events occur in the ascending aorta. 82 In particular, the rate of aortic dissection in pregnant women with ACTA2 mutations was significantly higher than similar unaffected cohorts. 83 A preclinical study suggests a plausible link between lactation-associated oxytocin release and aortic dissection. 84 As a result, women with heritable aortopathies contemplating pregnancy should be counseled on the risks and whether prophylactic surgical correction should be considered prior to pregnancy.
Smoking cessation
Tobacco use is an important risk factor for the development of aortic aneurysms, particularly AAAs. Several studies have found a dose–response relationship between the intensity of smoking and risk of AAAs.85,86 Further, a recent meta-analysis found a fivefold and twofold increase in the risk of AAAs among current and former smokers as compared to never smokers, respectively. 2 Tobacco use itself has been shown to have destructive effects on connective tissue and to increase the risk of aneurysmal rupture.87,88 In a recent study, cigarette smoking was associated with loss of immunogenic self-tolerance and induction of elastin-specific autoreactive T-cell responses in patients with thoracic aortic aneurysms and dissections. 89 Thus, smoking cessation should be strongly advocated in all patients with aortic aneurysms.
Conclusions and future directions
The rise of genome sequencing technologies in the past several decades has led to the identification of a number of heritable aortopathies and important insights that have improved understanding of the cellular basis for aneurysmal disease. Ongoing and future research will be important to personalize clinical management strategies based upon genetic lesions.
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: YK is supported by the National Heart, Lung & Blood Institute, American Venous Forum Foundation, Bo Schembechler Heart of a Champion Foundation, Frankel Cardiovascular Center, and has received cnsulting fees from Acer Therapeutics. The other authors have no relevant financial or COI disclosures.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.