
Abstract

Hyperactivating mutations in GNAQ have been discovered in a growing number of disorders including uveal melanoma, phakomatosis pigmentovascularis, port-wine birthmarks and Sturge–Weber syndrome (SWS).1,2 SWS, a sporadic neurocutaneous syndrome, is associated with facial port-wine birthmarks (PWBs), glaucoma, and a leptomeningeal angioma involving the brain. Patients present with seizures, stroke-like episodes, and cognitive impairment. The leptomeningeal vascular malformation consists of an increased number of tortuous abnormal blood vessels.
GNAQ codes for Gαq, part of the trimeric G protein complex associated with several G protein-coupled receptors. The R183Q mutation in GNAQ, which causes about 85% of SWS, 3 is predicted to impair auto-hydrolysis and deactivation of Gαq, and to cause constitutive hyperactivation of downstream pathways. Potential hyperactivated pathways include the Ras-Raf-MEK-ERK, 1 mTOR, 4 and YAP-HIPPO pathways. 5 The GNAQ mutation has been shown to be enriched in the endothelial cells of SWS leptomeningeal/superficial cortical brain samples. 6 We sought to determine the expression of Gαq and downstream phosphorylated extracellular signal-regulated kinase (p-ERK) in human SWS brain tissue to better understand how the somatic mutation results in a vascular malformation. CD34+ was selected as a marker of endothelial cells because, when labeling the inner layer of capillary-venous vessels, this marker is specific for endothelial cells, it does not also label other mature hematopoietic cells, and its role in differentiation, adhesion and migration is of potential interest in SWS angiogenesis. 7
Tissue sections from SWS fixed surgical brain specimens (n=12) and epilepsy control samples (n=8) were examined by standard histology (hematoxylin and eosin) and immunocytochemistry to determine the expression of CD34 (#ab81289; Abcam, Cambridge, MA), p-ERK and total ERK (#4370; Cell Signaling Technology, Danvers, MA) and mouse total ERK (#4696; Cell Signaling Technology), and for Gαq (#ab75825; Abcam). Sections were co-stained with DAPI (#8961; Cell Signaling Technology) for nuclei identification. ImageJ software (https://imagej.nih.gov/ij/) was used to determine the average intensity density of p-ERK and total ERK expression in endothelial cells of leptomeningeal blood vessels from representative images, and a Likert scale was used to quantify the brightness and continuity of CD34 staining (two fields of view/section). Only fixed tissue was available from these subjects, from which we were unable to obtain sufficient quality DNA to reliably genotype for the percentage of somatic R183Q GNAQ mutations.
An increased number of leptomeningeal blood vessels with thin or disorganized vessel walls were seen in the SWS samples. These overlay disorganized cortex, with increased numbers of vessels (Figure 1, panel B) and calcification in the cortical and sub-cortical layers compared to epilepsy controls (panel A). Most SWS samples (10/12) demonstrated leptomeningeal vessels with discontinuous CD34+ labeling (red, Figure 1, panel D) and CD34+ staining which was significantly less (median brightness score 1 in SWS leptomeningeal vessels (Figure 1, panel D) versus 3 in epilepsy control vessels (Figure 1, panel C); p=0.02). SWS samples had significantly higher p-ERK expression (normalized to total ERK; 79±25% vs 23±20%, p < 0.01; Figure 2, panels A and B, p-ERK – red and DAPI – blue) in the endothelial cells of leptomeningeal vessels than epilepsy control samples. Gαq expression was very low in the endothelial and perivascular cells of the leptomeninges of both control and SWS samples (Figure 2, panels C and D, respectively, and inserts showing blood vessels), while distinct Gαq expression was noted in non-vascular cells of the cortex (Figure 2, panels C and D, corner inserts) and subcortex (data not shown) of both.
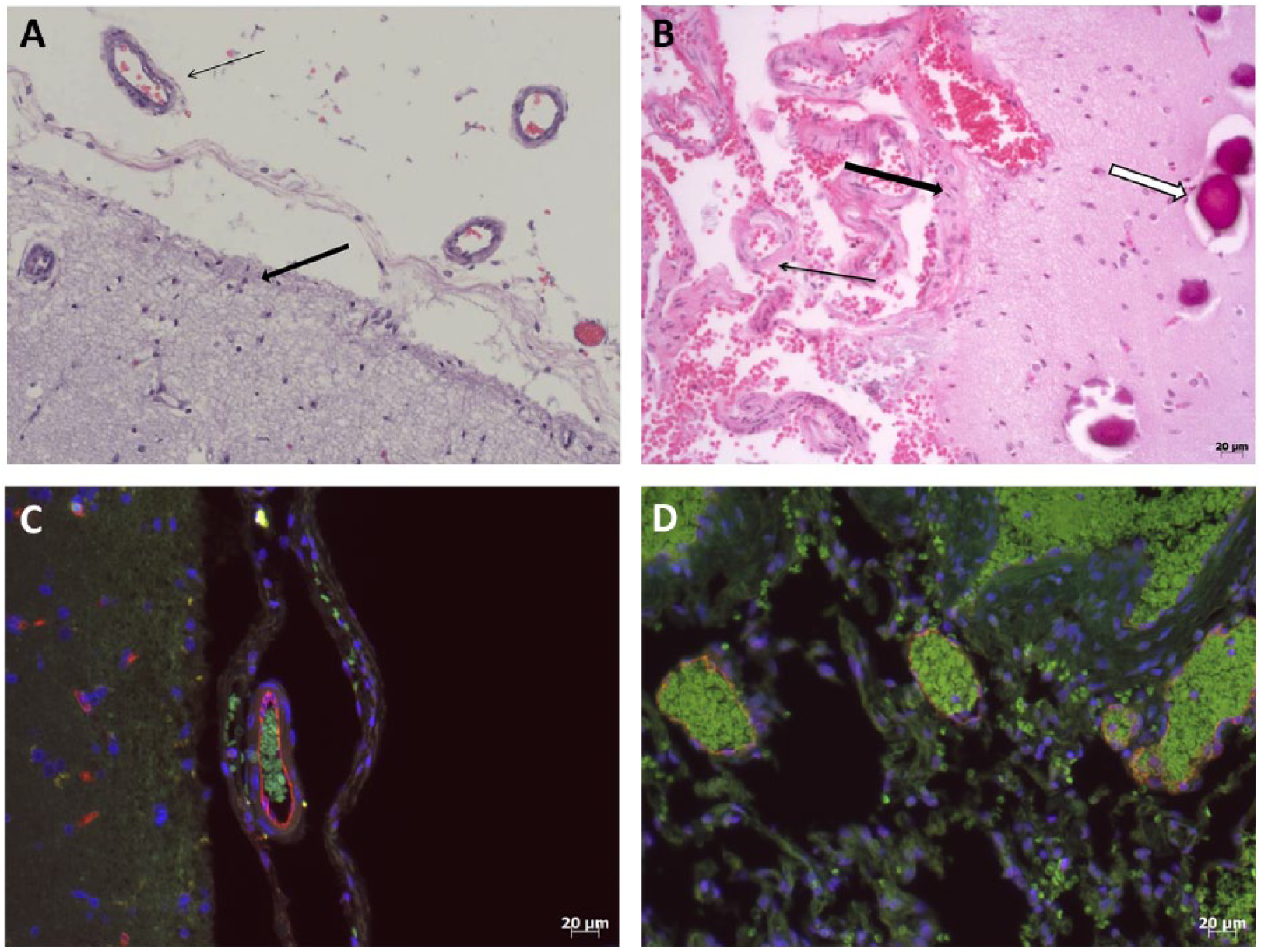
Surgical cortical brain samples stained with H&E, shown in grayscale, for both epilepsy control (panel A) and SWS (panel B) samples; leptomeningeal vessels (thin black arrows), as well as areas of cortex (thick black arrows), are indicated. The open arrow in panel B points to calcifications. Cortical sections were stained for CD34+ (red), α-tubulin (green), and DAPI (blue) for both epilepsy control (panel C) and SWS (panel D) samples.
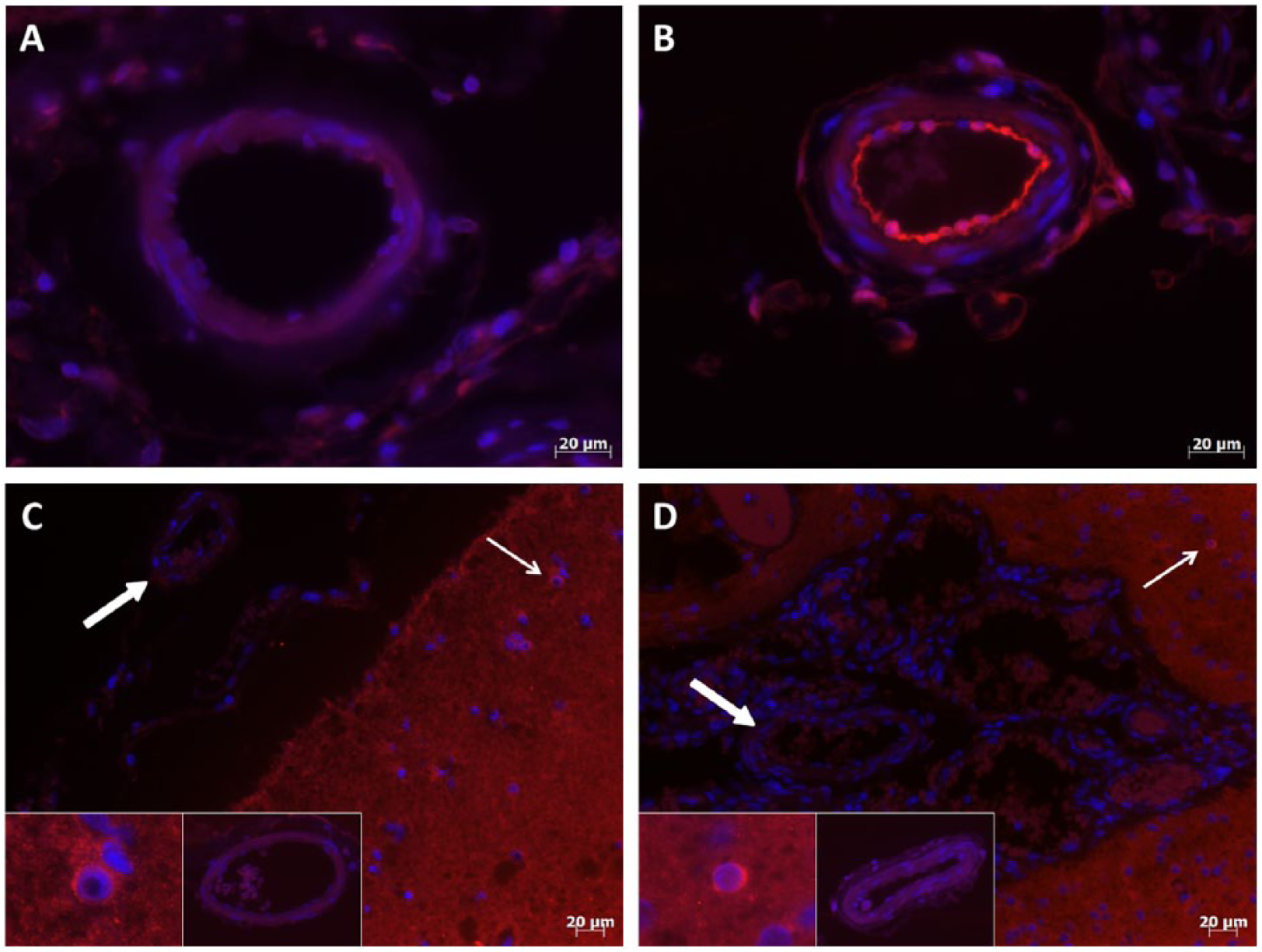
Sections were stained for p-ERK (red) and DAPI (blue) for SWS (panel B) and epilepsy control (panel A) samples. Phosphorylated-ERK expression was increased in the endothelial cells of abnormal leptomeningeal SWS blood vessels compared with epilepsy control. Additional sections (panels C and D) were stained for Gαq (red) and DAPI (blue); leptomeningeal vessels (thick arrows) of both epilepsy control (panel C) and SWS tissue (panel D) samples demonstrated similar low levels of Gαq labeling, while high levels of expression were seen in cells within the cortex (thin arrows). Inserts in panels C and D show examples of expression in cortical cells and leptomeningeal vessels taken at the same exposure.
Comati et al. reported in 2007 8 that the majority of SWS leptomeninges were thin-walled vessels of variable caliber, ectatic, CD34 labeled, and covered by a layer of smooth muscle/pericytes. They further noted that the abnormal leptomeningeal vessels did not have an internal elastic lamina, indicating that the vessel had venous characteristics. These SWS leptomeningeal endothelial cells had increased expression of vascular endothelial growth factor receptor (VEGFR)-1, VEGFR-2, hypoxia-inducible factor (HIF)1α and HIF2α. The authors suggested that VEGF released by the underlying hypoxic cortical cells may stimulate increased release of HIF1α by the abnormal leptomeningeal endothelial cells and further increases in VEGF expression. VEGF and VEGFR signal through Gαq, and therefore the hyperactivating R183Q mutation, may increase the expression of HIF1α through the Ras-Raf-MEK-ERK pathway, as suggested by our data. Constitutive hyperactivation of Gαq activity with mutant GNAQ does not necessarily impact levels of the expressed protein itself. Chronic hyperactivation of Gαq activity, due to decreased auto-hydrolysis of GTP to GDP, is predicted to result from the R183Q mutation, which impacts the auto-hydrolysis site. Compensatory changes in gene expression, in response to chronic constitutive activation of Gαq, might even lead to decreased Gαq protein levels, which would not be well measured by this approach (given the already low expression levels in control vessels). Further studies are needed of the Ras-Raf-MEK-ERK pathway to understand the mechanism of ERK hyperactivation by the mutant Gαq protein in SWS.
In port-wine birthmarks, increased endothelial p-ERK expression has been reported and may contribute to vascular structural and functional abnormalities. 9 Adult, hypertrophied port-wine birthmarks also demonstrate increased expression of other downstream kinases, suggesting that progressive hyperactivation of such pathways may contribute to worsening vascular ectasis over time. Increased VEGF may contribute to vascular hypertrophy in port-wine birthmarks. 10 We hypothesize that abnormal activation of these pathways results in abnormal gene expression, aberrant endothelial shape and CD34+ expression with breakdown of the endothelial barrier, and abnormal vascular remodeling leading to worsening perfusion deficits and neurologic dysfunction. CD34+ is normally found on small vessels and is lacking on larger vasculature; further study is needed to understand the relationship between decreased CD34+ endothelial expression and dilated leptomeningeal vessels. CD34+ endothelial cells are usually quiescent and are thought to be involved in blocking the migration and adhesion of lymphocytes. 7 A decrease in CD34+ staining suggests that the abnormal blood vessels may be more angiogenic and may more easily allow migration of inflammatory cells into the cortex. Interestingly, Gαq expression was very low in both SWS and the epilepsy control leptomeningeal vessel endothelial cells. Similar low expression of Gαq has been noted in endothelial cell cultures, with and without the R183Q mutation (data not shown). These low levels may, in part, contribute to the GNAQ mutation resulting in a vascular malformation rather than a tumor.
In summary, we show for the first time that phosphorylated ERK is increased in the endothelial cells of abnormal leptomeningeal vessels of SWS, and that this is associated with decreased expression of CD34+ and similar levels of endothelial Gαq. These results suggest that ERK inhibition may be a treatment target in SWS and provide a framework for beginning to understand the relationship between mutant Gαq and the vascular malformations in SWS. Further research with human tissue will be needed to validate and understand the new animal and in vitro models for SWS and port-wine birthmarks currently under development.
Footnotes
Acknowledgements
The BVMC Sturge–Weber syndrome Project Workgroup are: Karen L Ball, Sturge–Weber Foundation, Houston, TX, USA; Brian J Fisher, Sturge–Weber Foundation, Houston, TX, USA; Jim Koneig, Division of Neuroscience, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA; Douglas A Marchuk, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC, USA; Marsha A Moses, Vascular Biology Program, Boston Children’s Hospital, Boston, MA, USA, and Department of Surgery, Harvard Medical School, Boston Children’s Hospital, Boston, MA, USA; Jonathan Pevsner, Department of Neurology, Hugo W Moser Research Institute, Kennedy Krieger Institute, Baltimore, MD, USA, and Department of Psychiatry and Behavioral Sciences, Johns Hopkins School of Medicine, Baltimore, MD, USA.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by grants from the National Institute of Neurological Disorders and Stroke (NINDS) (National Institutes of Health (NIH) U54NS065705) (to Michael Lawton; Sturge–Weber Project to AMC). The Brain Vascular Malformation Consortium (U54NS065705) is a part of the NIH Rare Diseases Clinical Research Network (RDCRN), supported through the collaboration between the NIH Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Science (NCATS) and the NINDS. Brain tissue samples were obtained from the NIH Neuro BioBank, the Johns Hopkins Brain Tissue Bank, and the UCSF Brain Tissue Bank.