
Abstract
Obesity is associated with the development of vascular insulin resistance; however, pathophysiological mechanisms are poorly understood. We sought to investigate the role of WNT5A-JNK in the regulation of insulin-mediated vasodilator responses in human adipose tissue arterioles prone to endothelial dysfunction. In 43 severely obese (BMI 44±11 kg/m2) and five metabolically normal non-obese (BMI 26±2 kg/m2) subjects, we isolated arterioles from subcutaneous and visceral fat during planned surgeries. Using videomicroscopy, we examined insulin-mediated, endothelium-dependent vasodilator responses and characterized adipose tissue gene and protein expression using real-time polymerase chain reaction and Western blot analyses. Immunofluorescence was used to quantify endothelial nitric oxide synthase (eNOS) phosphorylation. Insulin-mediated vasodilation was markedly impaired in visceral compared to subcutaneous vessels from obese subjects (p<0.001), but preserved in non-obese individuals. Visceral adiposity was associated with increased JNK activation and elevated expression of WNT5A and its non-canonical receptors, which correlated negatively with insulin signaling. Pharmacological JNK antagonism with SP600125 markedly improved insulin-mediated vasodilation by sixfold (p<0.001), while endothelial cells exposed to recombinant WNT5A developed insulin resistance and impaired eNOS phosphorylation (p<0.05). We observed profound vascular insulin resistance in the visceral adipose tissue arterioles of obese subjects that was associated with up-regulated WNT5A-JNK signaling and impaired endothelial eNOS activation. Pharmacological JNK antagonism markedly improved vascular endothelial function, and may represent a potential therapeutic target in obesity-related vascular disease.
Introduction
The growing obesity epidemic has developed into a major health care problem globally, with currently 69% of the US population and over 2 billion people worldwide classified as overweight or obese.1,2 Premature heart disease and stroke are the leading causes of death in these populations, and clinical studies demonstrate that insulin resistance may be a key contributor to atherosclerosis in obesity.3,4 In particular, central adiposity with intra-abdominal accumulation of visceral fat has been linked to adipocytokine overproduction, insulin resistance, and cardiovascular risk; however, regulatory mechanisms that govern these pathogenic processes are poorly understood.5–10 Recent work from our group described non-canonical wingless-related integration site (WNT)5A signaling as a novel pro-inflammatory mediator central to mechanisms of obesity-related insulin resistance and endothelial dysfunction.11–13 WNTs play important roles in adipose tissue homeostasis, and combined animal and human studies have recently provided evidence that WNT5A can serve as a master regulator of inflammatory processes in a c-jun N-terminal kinase (JNK)-dependent manner. Non-canonical WNT signaling is of clinical interest, and implicated in several conditions including cancer, arthritis, and aging.14–16 Additionally, WNT5A is present in human atherosclerotic lesions, circulates in blood at higher levels in obese compared to lean persons, and low serum levels of Sfrp5 (a WNT5A antagonist) are linked with coronary disease.17–20 However, little is known about the role of WNT5A-JNK in mediating pathophysiology in the human vasculature. Thus, the goal of the current study was to characterize insulin-mediated vasodilator responses in human adipose tissue arterioles prone to endothelial dysfunction, and test the hypothesis that targeting WNT5A-JNK may reverse vascular insulin resistance associated with obesity and metabolic dysfunction.
Materials and methods
Study subjects
Obese men and women (body mass index (BMI) ⩾30 kg/m2, age ⩾18 years) were enrolled from the bariatric surgery program at Boston Medical Center (BMC). Non-obese men and women (BMI <30 kg/m2, age ⩾18 years) were recruited from elective abdominal surgery at BMC. Subjects with unstable medical conditions such as active coronary syndromes, congestive heart failure, systemic infection, acute illness, malignancy or pregnancy were excluded. The study was approved by Boston Medical Center Institutional Review Board and all subjects provided written informed consent. During a pre-surgical outpatient visit, clinical characteristics including blood pressure, heart rate, height, weight, BMI, and waist circumference were recorded. Fasting blood samples were drawn for biochemical analyses including lipids, glucose, insulin, homeostasis model assessment of insulin resistance (HOMA), glycosylated hemoglobin (HbA1c), and high-sensitivity C-reactive protein (hs-CRP) at the time of the pre-surgical visit. All biochemical analyses were performed by the Boston Medical Center clinical chemistry laboratory.
Adipose tissue collection and vessel preparation
Subcutaneous and visceral adipose tissue biopsies were collected intra-operatively during surgery. Subcutaneous adipose tissue was harvested from the lower abdominal wall and visceral fat obtained from the greater omentum. Adipose tissue specimens were placed immediately in cold HEPES buffer solution, pH 7.51 (American Bioanalytical, Natick, MA, USA). Small adipose arterioles (75–350 µm internal diameter) were carefully removed of surrounding fat and connective tissue. Arterioles were cannulated securely with glass micropipettes within an organ chamber as previously described.5,7 The chamber was then mounted onto a stage of an inverted microscope (200× magnification) and video camera monitor (model VIA-100; Boeckler Instruments, Inc., Tucson, AZ, USA) for vascular diameter measurements using videomicroscopy. Arterioles were pressure equilibrated and continuously perfused with Krebs buffer aerated with a gas mixture of 5% O2, 21% CO2 and 74% N2.
Assessment of adipose arteriolar function
The internal arterial diameter of each vessel was initially measured at a steady state followed by administration of endothelin-1 (ET-1, 2×10−6 M; Sigma-Aldrich, St Louis, MO, USA) to pre-constrict vessels to 50–70% of their internal diameter. Insulin-mediated, endothelial nitric oxide synthase (eNOS)-dependent vasodilation was determined by measuring the change in diameter in response to insulin (10−5 M; Sigma-Aldrich) at 5-minute intervals over a time course of 30 minutes (n=18 subcutaneous and n=33 visceral vessels from obese subjects, n=5 visceral vessels from non-obese subjects). 21 Vessel viability and maximum dilator capacity was examined using the endothelium-independent vasodilator papaverine (Pap, 2×10−4 M; Sigma-Aldrich). To assess the contribution of JNK to vascular insulin signaling, the JNK inhibitor (SP600125, 10−9 M; Sigma-Aldrich) was administered to the vessel chamber after initial quantification of insulin-induced vasodilation. The effect of JNK was measured on vasodilator response immediately following administration of the inhibitor (n=17 vessels from the visceral fat of obese subjects). Endothelium-independent vasodilation was determined using papaverine. Pharmacological agents were added directly to the external bathing solution of the organ chamber and the indicated concentration represents the final chamber molar concentration.
Endothelial cell isolation from whole adipose tissue
Subcutaneous adipose tissue biopsies were collected during bariatric surgery and placed immediately into cold DMEM (Gibco Life Technology, Grand Island, NY, USA) supplemented with penicillin, and streptomycin and 0.5% serum. Tissue was cut into small pieces, minced and digested in collagenase I (2.5 μg/ml; Sigma-Aldrich) for 1 hour in a 37°C water bath in a 100 rpm rotation and passed through a 70 μM filter to remove any remaining undigested tissue. Cells were then centrifuged at 600 rpm at 4°C for 10 minutes to separate adipocytes (top layer), and red blood cells were lysed using 1X RBC lysis buffer (R&D Systems, Minneapolis, MN, USA). The remaining cells were labeled with CD31 microbeads (Miltenyi Biotech, Auburn, CA, USA) before being loaded into the autoMACS Pro Separator. Isolated CD31+ endothelial cells were plated on fibronectin (Fisher Scientific, Pittsburg, PA, USA) coated eight-well chamber slides (BD Bioscience, San Jose, CA, USA). Cells were pre-treated with recombinant WNT5A (R&D Systems) for 48 hours and then serum starved (0.5% serum media) for 6 hours prior to a 30 minute insulin stimulation (10−7 M). Cells were then fixed immediately in 4% paraformaldehyde.
Endothelial cell protein expression by quantitative immunofluorescence
Stimulatory activation via phosphorylation of endothelial nitric oxide synthase (p-eNOS) at serine 1177 in response to insulin was assessed as previously described.11,21–23 Briefly, fixed endothelial cells were rehydrated with 50 mM glycine (Sigma-Aldrich), permeabilized with 0.1% Triton-X and blocked with 0.5% bovine serum albumin (BSA)/50 mM glycine. Slides were incubated for an hour at 37°C with primary antibodies against p-eNOS at serine 1177 (1:100 dilution; Abcam, Cambridge, MA, USA) and von Willebrand factor (vWF, 1:300 dilution; Dako, Carpinteria, CA, USA) to select endothelial cells. Analogous Alexa Fluor-488 and Alexa Fluor-594 antibodies (1:200 dilution; Invitrogen, Carlsbad, CA, USA) were used for the secondary antibodies. Cells were mounted under glass coverslips with Vectashield (Vector Laboratories, Burlingame, CA, USA) containing DAPI to identify nuclei. Slides were imaged using a fluorescent microscope (20× magnification; Nikon Eclipse TE2000-E) and digital images were captured using a Photometric CoolSnap HQ2 Camera (Photometrics, Tucson, AZ, USA). Exposure time was kept constant and fluorescent intensity (corrected for background fluorescence) was quantified by NIS Elements AR Software (Nikon Instruments, Melville, NY, USA). Fluorescent intensity was quantified in 20 cells from each depot/subject and averaged. Batch-to-batch staining variability was controlled for by simultaneously staining human aortic endothelial cells (HAEC). Fluorescence intensity for each sample was normalized to the intensity of HAEC as previously validated.7,23,24
Adipose tissue gene expression
Adipose tissue was collected, placed in Allprotect Tissue Reagent (Qiagen, Germantown, MD, USA) and stored at −80°C until further processing. Total RNA was extracted from adipose tissue samples using Qiagen RNeasy Lipid Tissue Mini Kit and the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) for cDNA synthesis. Quantitative real-time polymerase chain reaction (PCR) was performed using TaqMan gene expression assays (Applied Biosystems). Data normalized to GAPDH were compared using the ΔΔct method and expressed as fold-induction in gene expression in visceral adipose tissue compared to subcutaneous fat.
Western immunoblot analyses
Proteins were extracted from whole adipose tissue as previously described. 23 Fat was homogenized in liquid nitrogen. Ice-cold 1X lysis buffer (Cell Signaling, Danvers, MA, USA) was supplemented with protease inhibitor cocktail and phosphatase inhibitor II and III (Sigma-Aldrich) and added to the homogenate. Samples were assayed for protein content using Bradford’s method. A total of 30 µg of protein was subjected to electrophoresis in SDS-polyacrylamide gel under reducing conditions and blotted to nitrocellulose membrane using the Bio-Rad Transblot Turbo Transfer System (Hercules, CA, USA). The membranes were blocked in 5% BSA, 0.1% Tween-20 in Tris-buffered saline (TBST) for 1 hour at room temperature and then incubated overnight at 4°C with primary anti-human antibodies (1:1000). Membranes were washed with TBST and incubated with horseradish peroxidase-conjugated secondary anti-rabbit IgG (R&D Systems) for 1 hour at room temperature; immune complexes were detected with the enhanced chemiluminescence ECL detection system (Bio-Rad). The ImageQuant LAS 4000 biomolecular imaging system (GE Healthcare, Pittsburg, PA, USA) was used to perform densitometric analyses of protein bands. Proteins of interests were JNK and pJNK, phosphorylated at Thr183/Tyr185 (Cell Signaling). The intensity of bands for each protein was normalized to the loading control beta-actin band (Cell Signaling).
Statistical analysis
Differences in clinical characteristics and baseline vascular parameters between groups were examined by independent t-tests or Mann–Whitney–Wilcoxon for continuous variables, as appropriate. Kolmogorov-Smirnov tests and histograms were used to determine whether continuous variables were normally distributed or skewed. Chi-squared tests were used to determine differences for categorical variables. Repeated measures (RPM) analysis of variance (ANOVA) was used to compare depot-specific vascular time-response curves to pharmacological agents. Given the nature of the study design that was intended to explore novel mechanisms of vascular dysfunction in obesity, statistical correction for multiple testing was not performed. Differences in gene expression, protein phosphorylation with/without insulin stimulation and group differences between subcutaneous and visceral depots were analyzed using paired Student’s t-tests. Associations between vascular dilator responses defined by time-response area under the curve (AUC), adipose tissue gene expression, and clinical data were examined using Spearman’s rank correlation. Statistical significance was defined as p<0.05. All data are expressed as mean ± SD, unless otherwise indicated. All data were analyzed using SPSS for Windows, version 19.0 (IBM Corp., Armonk, NY, USA).
Results
Clinical characteristics
A total of 51 arterioles were harvested from subcutaneous (n=18) and visceral (n=33) adipose depots in 43 severely obese subjects undergoing planned bariatric surgery at Boston Medical Center. Additionally, five metabolically healthy non-obese subjects provided adipose tissue microvessels (n=5 visceral arterioles) removed during their elective abdominal surgical procedures. The clinical characteristics of subjects who provided adipose tissue arterioles are displayed in Table 1. As expected, obese subjects were metabolically abnormal with greater prevalence of insulin resistance and systemic inflammation. Within the obese cohort, no significant differences in clinical parameters were observed between subjects who provided blood vessels from subcutaneous or visceral adipose tissue depots. The average internal resting diameter of arterioles was 177±49 µm isolated from visceral fat and 161±58 µm (p=0.10) from subcutaneous fat of the obese subjects, with mean pre-constriction diameters of 65±13% and 67±6% (p=0.76), respectively. Arterioles from lean subjects were pre-constricted to 66±9% of the resting diameter, which was on average 187±80 µm in diameter (p=0.90 vs visceral arterioles of obese).
Study population characteristics.
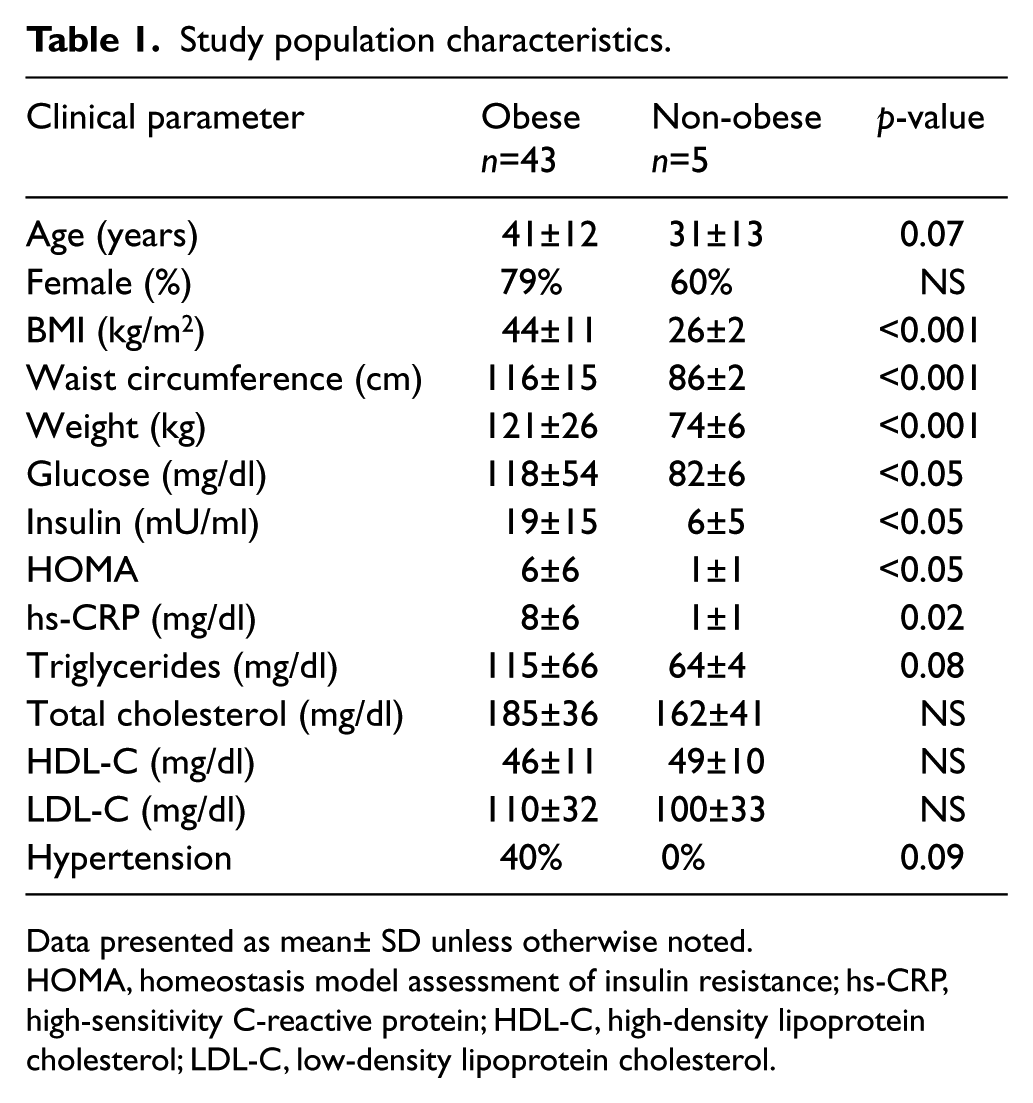
Data presented as mean± SD unless otherwise noted.
HOMA, homeostasis model assessment of insulin resistance; hs-CRP, high-sensitivity C-reactive protein; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol.
Adipose depot-specific vascular responses
Insulin-mediated endothelium-dependent vasodilation was assessed by dilator response curves to 10−5 M insulin quantified over a period of 30 minutes. As shown in Figure 1A, we observed significant impairment of insulin-stimulated vasodilation in visceral compared to subcutaneous adipose tissue arterioles in obese subjects (p<0.001). Responses to papaverine were intact, indicating that the impairment developed at the level of the vascular endothelium. Depot-specific differential responses to insulin were also observed in a subset of nine subjects that had provided paired subcutaneous and visceral microvessels from the same individual (p<0.001, data not displayed). Insulin-mediated vasodilation (AUC) in vessels from visceral fat of diabetic obese subjects correlated negatively with weight (r = −0.9, p=0.01) and BMI (r = −0.8, p=0.04). Interestingly, as displayed in Figure 1B, insulin-induced responses appeared to be preserved in microvessels isolated from the visceral fat of healthy non-obese subjects, demonstrating that impairment in visceral arteriolar function develops specifically as a function of obesity.
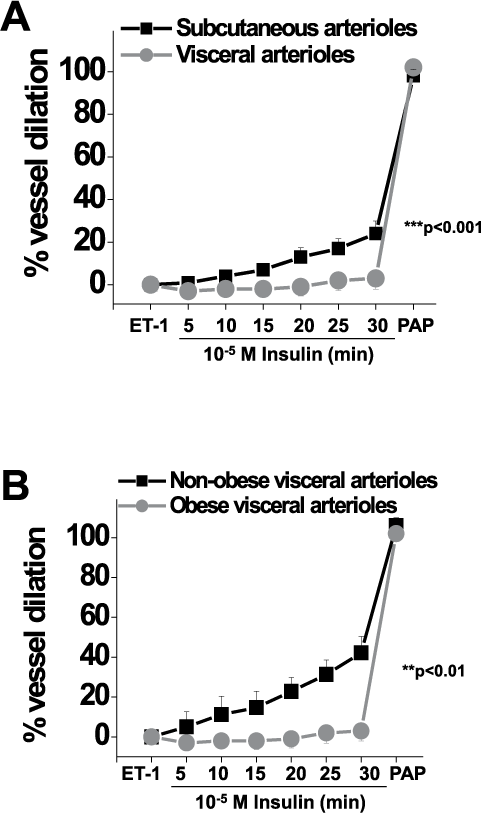
Endothelium-dependent, insulin-mediated vasodilation of adipose arterioles. (A) Endothelium-dependent, insulin-mediated vasodilation was severely impaired in visceral compared to subcutaneous adipose tissue arterioles of obese subjects (n=18 subcutaneous and n=33 visceral vessels, p<0.001 by repeated measures (RPM) ANOVA). (B) Endothelium-dependent, insulin-mediated vasodilation was preserved in visceral arterioles of non-obese subjects (n=5 vessels) compared to obese subjects (n=33 vessels, p<0.01 by RPM ANOVA). Data presented as mean ± SEM. (ET-1, endothelin-1; PAP, papaverine.)
Association of the insulin signaling transcriptome with WNT5A-JNK expression
In 27 obese subjects, we examined mRNA transcripts of components of the insulin signaling cascade and observed down-regulation of insulin receptor (INSR), insulin receptor substrate 1 (IRS1), protein kinase b (AKT), and 3-phosphoinositide-dependent protein kinase 1 (PDPK1) in visceral compared to subcutaneous adipose tissue (Table 2). Consistent with previous studies from our group, 12 WNT5A expression was increased in visceral fat whereas prototypical canonical WNTs (WNT3A and WNT10) were not differentially expressed. The non-canonical co-receptors receptor tyrosine kinase-like orphan receptor 1 (ROR1) and ROR2 were also markedly upregulated in visceral fat. We observed significant negative correlations between ROR1 and ROR2 transcripts with expression of AKT, INSR, and IRS1 within omental fat, and similar associations were also noted in subcutaneous fat (Table 3). Activation of JNK downstream from WNT5A impairs insulin sensitivity and promotes adipose inflammation in obesity.11–13,25,26 In line with this, we observed a significant upregulation of JNK activation (phosphorylation) in visceral compared to subcutaneous fat (Figure 2A; n=19, p<0.05), which correlated with plasma hs-CRP (Figure 2B; r=0.72, p<0.05). Collectively, these data suggest that elevated WNT5A-ROR-JNK signaling within adipose tissue in obesity is associated with down-regulation of the insulin signaling pathway, and that this signaling may contribute to mechanisms of obesity-related insulin resistance.
Gene expression in visceral compared to subcutaneous adipose tissue.
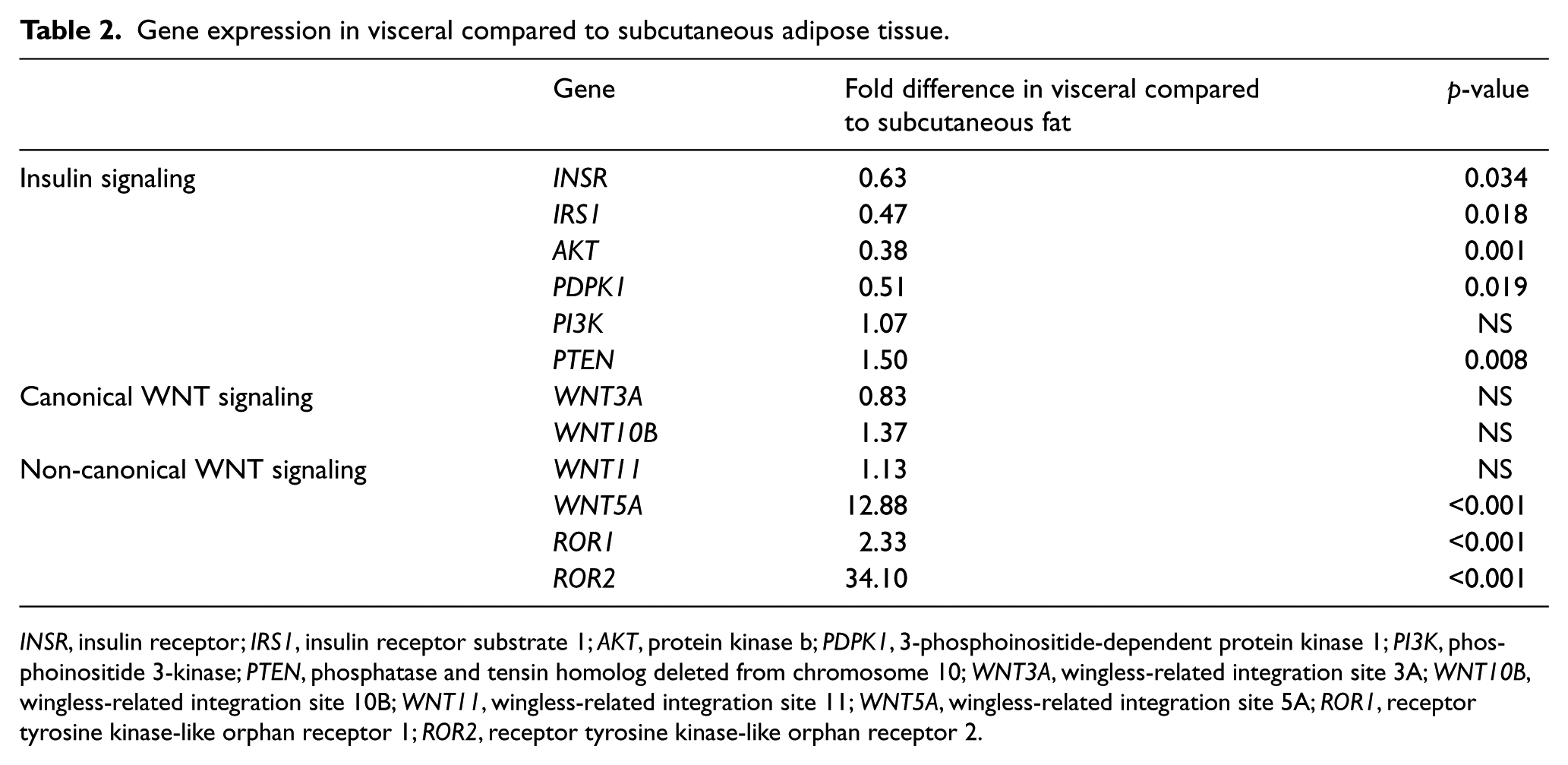
INSR, insulin receptor; IRS1, insulin receptor substrate 1; AKT, protein kinase b; PDPK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog deleted from chromosome 10; WNT3A, wingless-related integration site 3A; WNT10B, wingless-related integration site 10B; WNT11, wingless-related integration site 11; WNT5A, wingless-related integration site 5A; ROR1, receptor tyrosine kinase-like orphan receptor 1; ROR2, receptor tyrosine kinase-like orphan receptor 2.
Correlations between genes relevant to WNT5A-ROR and insulin signaling in adipose tissue.
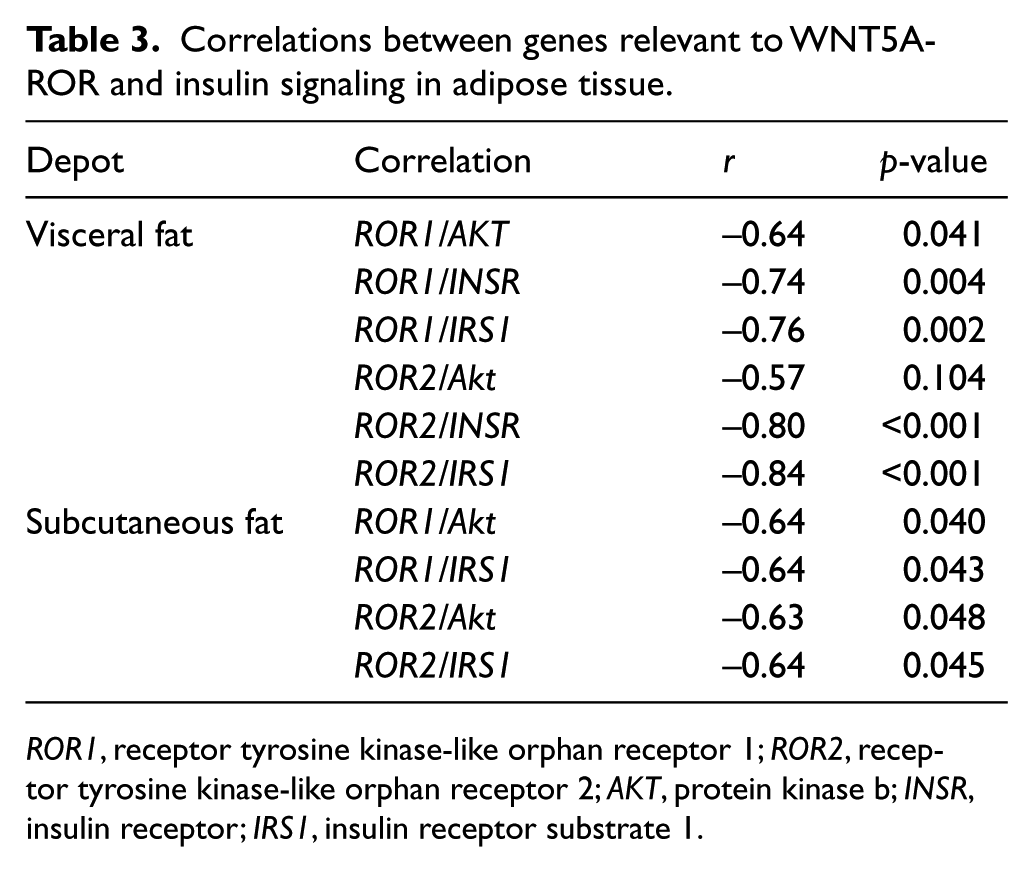
ROR1, receptor tyrosine kinase-like orphan receptor 1; ROR2, receptor tyrosine kinase-like orphan receptor 2; AKT, protein kinase b; INSR, insulin receptor; IRS1, insulin receptor substrate 1.
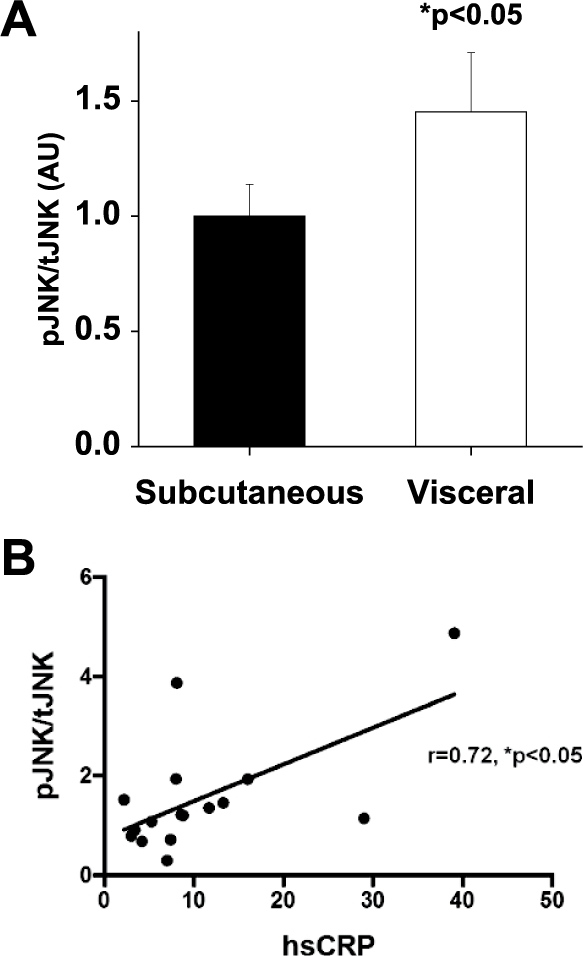
Activation of JNK in adipose tissue of obese subjects. (A) Quantification of the ratio of phosphorylated-JNK (activated, pJNK) to total-JNK (tJNK) normalized to β-actin in subcutaneous and visceral fat (n=19, p<0.05). Data presented as arbitrary units (au), subcutaneous depot indexed to 1. (B) Basal pJNK/tJNK protein expression in visceral adipose tissue correlated significantly with systemic hs-CRP in obese subjects (n=16, p<0.05). Data presented as mean ± SEM.
WNT5A-JNK modulates insulin-mediated vasodilation and eNOS activity
To determine the role of JNK in vascular insulin resistance, we assessed vascular responses to the pharmacological JNK inhibitor SP600125 (10−9 M).11,12 As shown in Figure 3A, treatment with SP600125 improved insulin-mediated vasodilation in visceral adipose arterioles by nearly sixfold (n=17, p<0.001). The whole vessel physiological data with suspended arterioles suggest that vasomotor impairment occurs as a result of endothelial dysfunction since vessels exhibit preserved responses to the endothelium-independent vasodilator papaverine. To provide further evidence for cell-autonomous functional defects and elucidate molecular mechanisms at the level of the endothelium, we developed methods to isolate endothelial cells from human adipose tissue and characterized their functional properties using quantitative immunofluorescence of eNOS phosphorylation.7,11,21–23 For this protocol, eNOS phosphorylation was examined at serine 1177 (p-eNOS), the commonly reported index of eNOS stimulatory activation in endothelial cells.27,28 Thus, p-eNOS was examined in isolated endothelial cells at basal levels and after insulin stimulation ± recombinant WNT5A (rWNT5A, 5×10−7 M). As shown in Figure 3B, treatment with rWNT5A did not alter basal levels of p-eNOS; however, it abolished insulin-mediated p-eNOS activation in subcutaneous endothelial cells (n=7, p<0.05), recapitulating the severe insulin-resistant state seen in visceral depots. Collectively, these data suggest that WNT5A-JNK activation contributes to vascular insulin resistance in human obesity, in part, owing to impaired eNOS activation and nitric oxide bioaction.
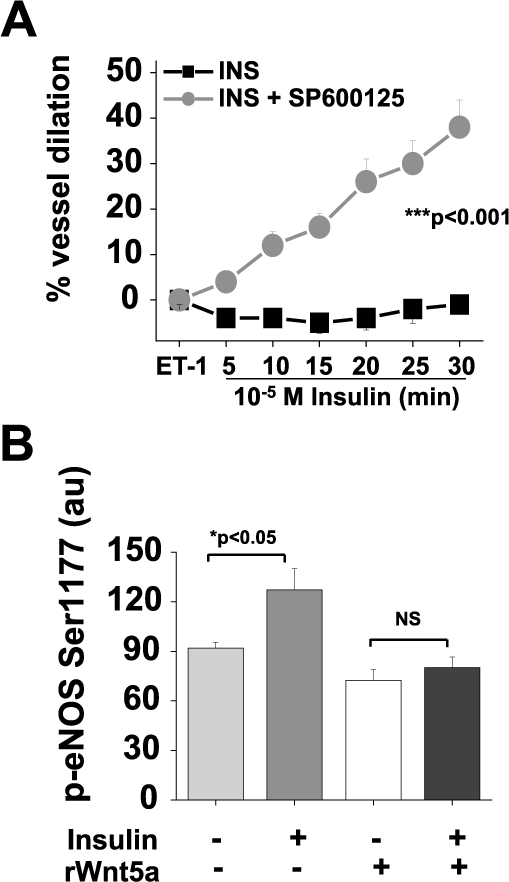
Effect of WNT5A-JNK signaling on insulin-mediated vasodilation and activation of eNOS. (A) SP600125 (10−9 M) treatment of arterioles from visceral fat significantly improved insulin-mediated vasodilation by sixfold (n=17 vessels, p<0.001 by repeated measures (RPM) ANOVA). Data presented as mean ± SEM. (B) Phosphorylation of eNOS at serine 1177 in response to insulin was attenuated by rWNT5A treatment in endothelial cells isolated from subcutaneous fat of obese subjects (n=7, p<0.05). Data presented as arbitrary immunofluorescence units (au) and displayed as mean ± SEM.
Discussion
In the present study, we observed profound impairment in insulin-mediated, endothelium-dependent vasodilation of arterioles isolated from visceral compared to subcutaneous adipose tissues of severely obese humans. Non-canonical WNT5A expression is elevated in visceral fat, which correlates negatively with the expression of components of the insulin signaling cascade. Moreover, we observed increased p-JNK protein expression in omental fat which was associated with elevated plasma hs-CRP levels, and pharmacological JNK antagonism improved vascular insulin resistance in dysfunctional microvessels. Taken together, our findings suggest that modulation of the WNT5A-JNK pathway may represent a target in the restoration of vascular insulin sensitivity.
It is widely accepted that central obesity and accumulation of intra-abdominal visceral fat are strongly associated with the development of insulin resistance and cardiovascular disease.5–7,10,12,29–31 Adipose tissue dysfunction and over-production of pro-atherogenic cytokines contribute to a low-grade systemic inflammatory state which has been strongly implicated in the pathogenesis of cardiometabolic disease; however, regulatory mechanisms are poorly understood and treatment targets remain elusive. We recently described the up-regulation of a unique pro-inflammatory pathway involving non-canonical WNT5A signaling linked to insulin resistance and vascular disease in obesity.12,13,32 WNT proteins evolved with the very first animals, and their complex physiology consists of secreted signaling molecules that play fundamental roles in regulating differentiation and body axis patterning during embryonic development. WNT signaling pathways are generally classified as either canonical (β-catenin-dependent), typified by WNT1, WNT3A, and WNT10B, or non-canonical (β-catenin-independent) mainly WNT5A and WNT11. WNTs share the same Frizzled (Fzd) receptors, but associate with specific co-receptors that mediate canonical (LRP 5 and 6) or non-canonical (ROR 1 and 2) signaling. Recent clinical data have implicated aberrant WNT signaling in the pathogenesis of adult diseases such as cancer, arthritis, and cardiovascular disease.14,16
In the current study, we found significant up-regulation of WNT5A in visceral compared to subcutaneous fat, consistent with our previous observation. 12 Even more striking was marked over-expression of WNT5A co-receptors, particularly ROR2, which exhibited a remarkable 34-fold higher expression in visceral fat. This is in contrast to the typical observed range of differential up-regulation of adipocytokines (generally less than fivefold), 5 suggesting that WNT5A may represent a key regulator of immune responses in dysfunctional human fat. Among other potential signaling actions, WNT5A-ROR2 is known to activate JNK33–35 and experimental studies have identified the JNK pathway as a key contributor to the development of insulin resistance, adipose tissue dysfunction, and atherosclerosis.13,25,26,36–38 Clinical studies demonstrate increased JNK in atherosclerotic lesions and arterioles isolated from the visceral fat of obese subjects.11,39–41 Additionally, we recently reported that endothelial cells harvested from the forearm circulation of obese type 2 diabetics display over-activation of WNT5A/JNK signaling, impaired eNOS phosphorylation, and reduced nitric oxide production, providing evidence for a systemic disease process and parallel abnormalities in both adipose and peripheral vascular beds. Our salient observations that JNK inhibition significantly improves insulin-mediated vasodilation, and that endothelial cells exposed to recombinant WNT5A develop insulin resistance, provide mechanistic support that WNT5A-JNK signaling is involved in the regulatory control of vascular phenotype.
Endothelial dysfunction represents a key step in the evolution of cardiovascular disease; thus, signaling pathways that regulate endothelial function are pivotal for understanding atherosclerosis initiation and progression. Insulin modulates vascular tone and blood flow largely through activation of endothelial NO synthase (eNOS) by binding to its IRS1-linked receptor with subsequent phosphorylation and activation of eNOS at Ser1177 via PI3K/Akt. Defective insulin signaling has been associated with vasoconstriction and ischemia, and endothelium-specific deletion of the insulin receptor in animal models leads to atherosclerosis.42–45 The degree of vascular insulin resistance in some cases was remarkable in the visceral microenvironment, with some arteriolar segments exhibiting paradoxical vasoconstriction indicative of severe endothelial dysfunction. In contrast, insulin-induced vasodilation in the visceral domain of metabolically healthy non-obese subjects were preserved, demonstrating that vascular dysfunction occurs as a function of obesity rather than a de facto pathophysiological property of visceral fat, and this observation is in line with other clinical studies that studied lean subjects.23,39,46
Our finding that non-canonical WNT signaling correlates negatively with several components of the insulin signaling cascade is an extension of our prior publications in both animal models (i.e murine knockout) and human tissue,11–13 and provides further evidence for a regulatory role of WNT5A in metabolic dysfunction. Our experimental model utilizing videomicroscopy for studying arterioles provides the ability to directly probe pathophysiology in intact whole segments of blood vessels removed from living subjects and gain insight into pathways that are differentially altered in disease conditions. Assessment of adipose arteriolar function has been shown to correlate with in vivo measures of systemic endothelial function and cerebrovascular responses within an individual, and associate with cardiovascular risk factors including hypertension, smoking, diabetes, and inflammation.47,48 Thus, characterization of microvascular responses in obesity may provide translational application to mechanisms relevant to systemic blood vessels.
Limitations
Our current study has several potential limitations. First, this study population comprised severely obese subjects referred for bariatric surgery and findings may not be applicable to individuals with milder degrees of obesity. However, this limitation is counterbalanced by the ability to study the pathophysiology of intact human blood vessels which would otherwise be very difficult. Second, the experimental model involved ex vivo manipulation that may not fully recapitulate in vivo conditions, although studies were performed immediately under conditions that approximate the physiological state. Third, the majority of study participants were women, which reflects the general clinical practice and known gender differences in populations that seek weight loss treatments.49,50 Fourth, we examined responses within the adipose tissue microvasculature which may not reflect processes at the level of systemic conduit vessels or coronary circulation. Fifth, we did not characterize WNT5A-JNK signaling in the visceral microenvironment of non-obese subjects; however, previous studies have shown WNT5A and JNK expression to be lower in visceral fat and the local adipose microvasculature of lean individuals.17,39 Lastly, we provided evidence for a link between vascular insulin resistance and WNT5A-JNK signaling within our obese cohort, but cannot exclude other relevant signaling pathways that may function upstream from JNK activation.
Conclusions
In conclusion, we observed profound vascular insulin resistance in the visceral adipose tissue arterioles of obese subjects that was associated with upregulated WNT5A-JNK signaling and impaired endothelial eNOS activation. Pharmacological JNK antagonism markedly improved vascular endothelial function, and may represent a potential therapeutic target in treating obesity-related vascular disease.
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Apovian has participated on advisory boards for Amylin, Merck, Johnson & Johnson, Arena, Nutrisystem, Zafgen, Sanofi-Aventis, Orexigen, EnteroMedics, GI Dynamics, Scientific Intake, Gelesis and Novo Nordisk. Dr Apovian has received research funding from Eli Lilly, Amylin, Aspire Bariatrics, GI Dynamics, Pfizer, Sanofi-Aventis, Orexigen, MetaProteomics, Takeda, the Dr Robert C and Veronica Atkins Foundation, MYOS Corporation and Gelesis. She has also participated on the Takeda Speakers Bureau for the medication Contrave. Currently, Dr Apovian owns stock in Science-Smart LLC. Dr Walsh has received research funding from Takeda Pharmaceuticals.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Gokce is supported by National Institutes of Health (NIH) grants HL081587, HL114675, and HL126141. Dr Walsh is supported by NIH grants HL120160, HL126141, HL131006 and HL132564. Dr Karki is supported by the NIH T32 HL07224. Dr Hamburg is supported by NIH grants HL81587 and HL11539. Dr Bretón-Romero is supported by the American Heart Association Postdoctoral Fellowship 16POST27260178.