
Abstract
Mutations of the ACTA2 gene, which encodes the smooth muscle cell-specific isoform of α-actin protein, have recently been found to be among the most common genetic abnormalities observed in patients with familial thoracic aortic aneurysms/dissection (TAAD). Other reported vascular manifestations caused by these mutations include premature coronary artery disease and stroke. We report a young adult who presented with an acute brachial artery occlusion and was subsequently found to have aortopathy and an ACTA2 mutation. This expands the spectrum of vascular disease associated with ACTA2 mutation to include acute limb ischemia.
Case presentation
A 17-year-old female patient presented to the emergency department with acute right arm ischemia. A CT scan revealed an acute occlusive arterial thrombus in the right brachial artery (Figure 1). Diffuse thoracic large vessel ectasia was also present; the aortic root and ascending aorta measured 4.1 cm and 3.8 cm in diameter, respectively (Figure 2). The transverse arch, abdominal aorta, common carotids and subclavian arteries were also mildly dilated. No mural thrombus was visualized. There was no evidence of tortuosity involving the proximal portions of the carotid and vertebral arteries. An urgent right brachial embolectomy was performed. Intra-operative angiography showed that the proximal axillary artery was dilated but there was no evidence of dissection. The brachial artery was not stenotic and did not appear to be chronically diseased. Anticoagulation with intravenous fractionated heparin followed by warfarin was prescribed postoperatively. Pathology showed the explanted thrombus to have no unusual features. The patient had a history of remote patent ductus arteriosus (PDA) ligation at the age of 6 months. The patient had been previously diagnosed in childhood with possible ‘isolated partial aniridia’ (partial absence of the iris). The patient was a non-smoker and was not using oral contraception. She had no history of cerebrovascular disease. The family history was negative for hypercoagulable states or aortopathy. On exam, she had no dysmorphic features. Trans-esophageal echocardiography (TEE) confirmed aortic root and ascending aorta dilatation. The aortic valve was trileaflet. There was no patent foramen ovale. Work-up by rheumatology including C-reactive protein, ESR (erythrocyte sedimentation), rheumatoid factor, ANA (antinuclear antibody), c-ANCA (anti-neutrophil cytoplasmic antibody), p-ANCA and syphilis serology were negative. Extensive investigations arranged by the thrombosis service for hypercoagulable state, including factor VIII, protein C and S, antithrombin, activated protein C, lupus anticoagulant, B2GP1 IgG and IgM, anticardiolipin IgG and IgM, homocysteine, and factor V Leiden, were negative. Outpatient genetic testing was negative for Loeys-Dietz (TGFBR1 and TGFBR2), Marfan (FBN1) and Ehlers-Danlos (COL3A1) syndromes. Testing for MYH11 and SLC2A10 mutations were negative. Serial TTEs revealed progressive dilatation of the aortic root and ascending aorta.
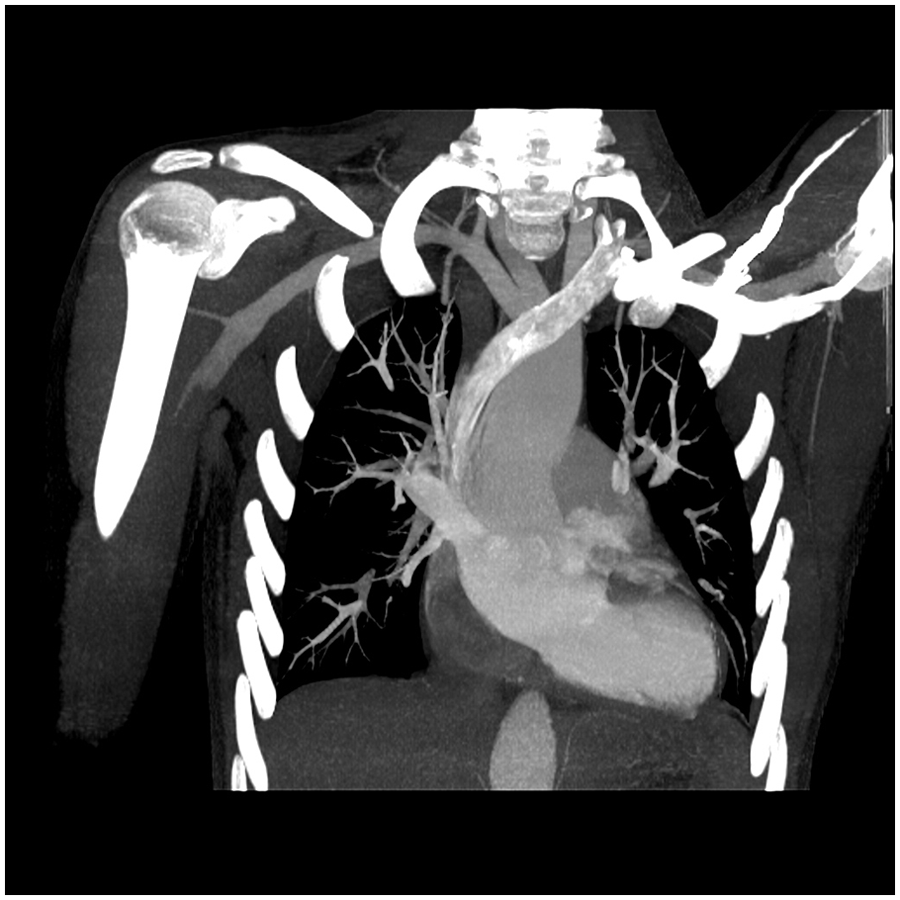
CT angiogram demonstrating an intra-arterial filling defect consistent with occlusive thrombus filling the lumen of the right brachial artery.
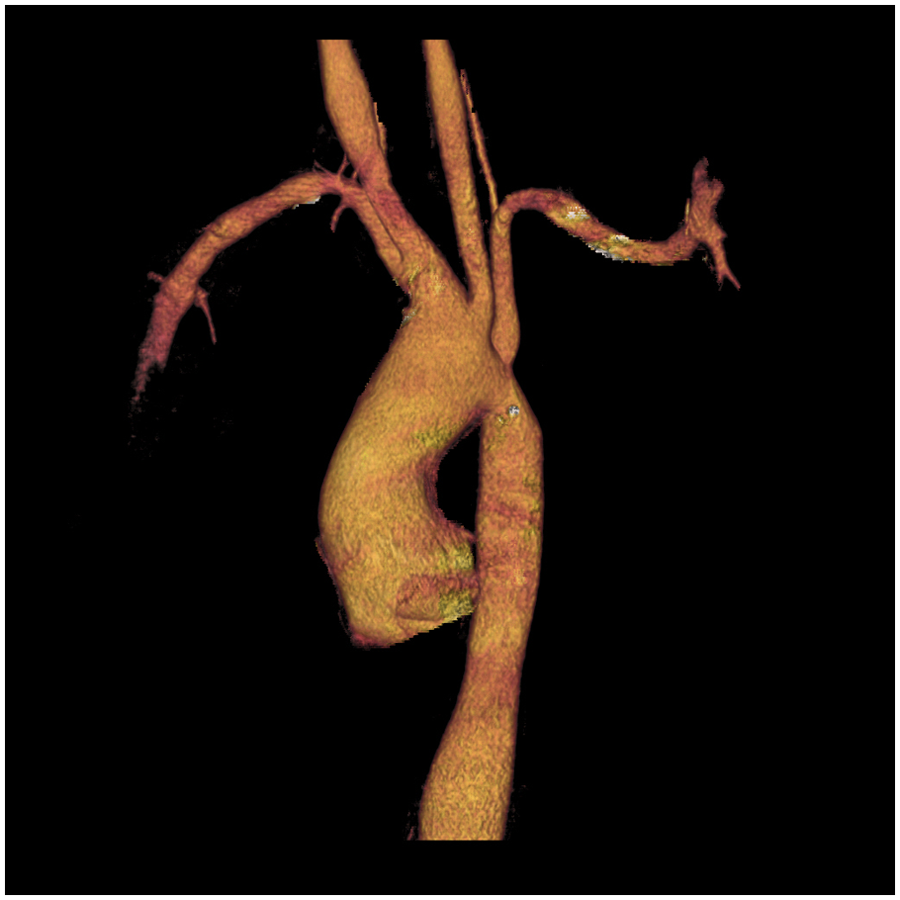
A 3D reconstructed CT image demonstrating aortic root, ascending aorta and abdominal aortic dilatation.
At 19 months after initial presentation, her aortic root and ascending aorta measured 4.7 cm (indexed to body surface area = 3.1 cm/m2). A MRI confirmed these measurements. Elective aortic root replacement was recommended and she underwent a David-1 valve sparing aortic root replacement with aortic valve repair. Histological examination of the aorta showed severe medial degenerative changes, ground substance accumulation and patchy moderate elastic fragmentation (Figure 3). No evidence of aortitis or dissection was seen.

Histopathology of the excised aorta demonstrating significant medial degenerative changes including elastic fragmentation and collections of glycosaminoglycan ground substances (hematoxylin phloxine saffron ×200).
In view of the childhood diagnosis of ‘isolated partial aniridia’, the patient was referred for ophthalmological assessment. Clinical findings of congenital aniridia were absent. The anterior segment examination was normal except for the irides, which were hypoplastic and fixed with the pupils mid-dilated (Figure 4). The pupils were completely unreactive to light, accommodation, and pharmacologic manipulation. Fundus examination revealed prominent retinal vasculature tortuosity but no foveal hypoplasia (Figure 5).
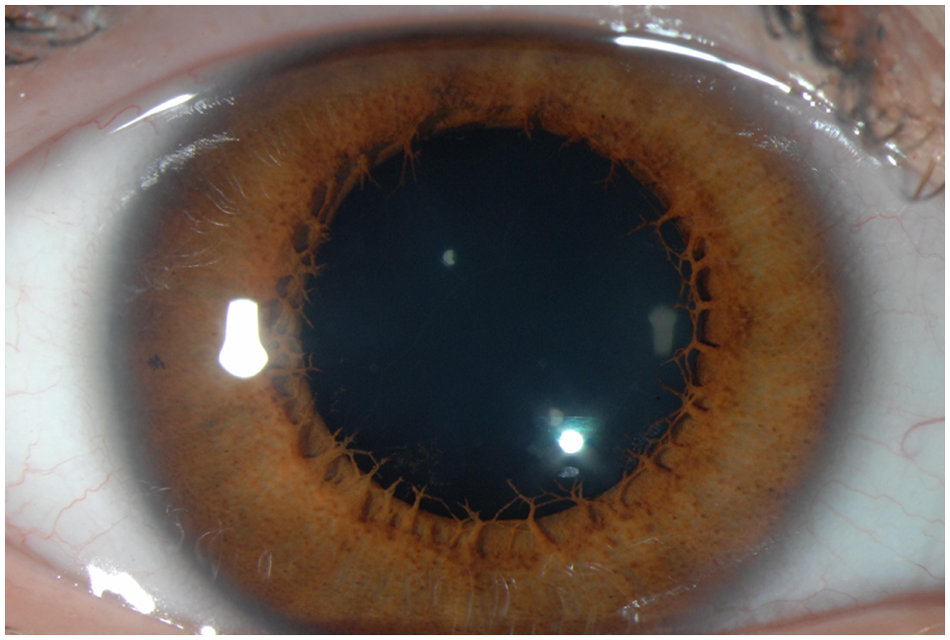
Anterior segment photograph of the eye, demonstrating congenital mydriasis.
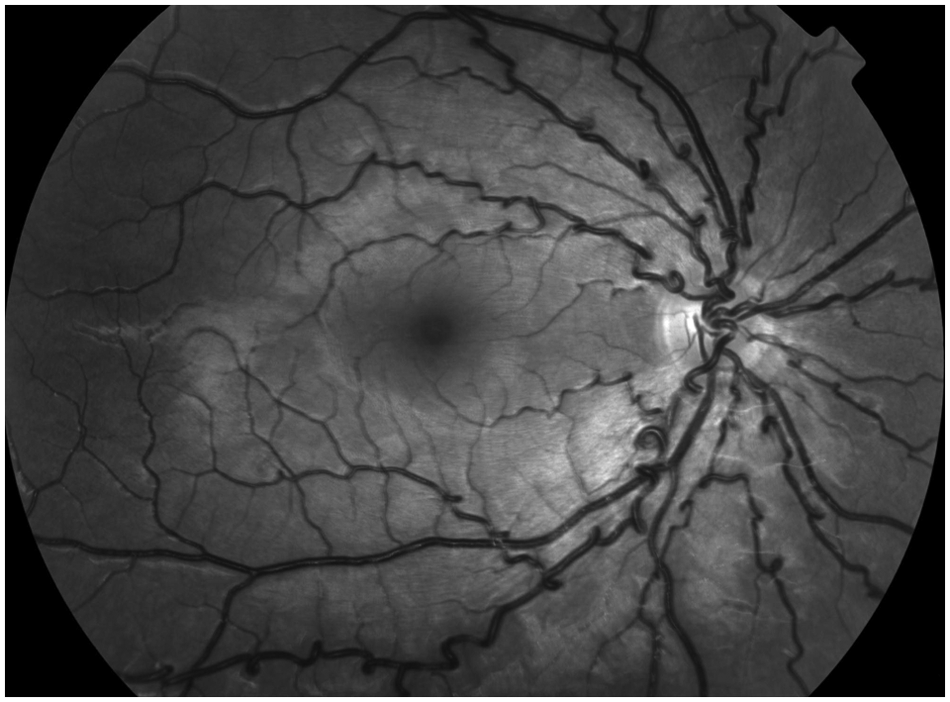
Fundus photograph of the eye, revealing prominent retinal vascular tortuosity.
The possibility of a mutation of the smooth muscle cell (SMC)-specific isoform of α-actin (ACTA2) was raised as a unifying diagnosis of her systemic and ocular findings. Genetic testing revealed a heterozygous c.536G>A transition exon 6 mutation (R179H) of the ACTA2 gene converting a codon for arginine (CGT) to a codon for histidine (CAT) (Connective Tissue Gene Tests, Allentown, PA, USA).
Discussion
There has been significant progress in the last few years in the understanding of the genetics of thoracic aortic aneurysm/dissection (TAAD) syndromes. Indeed, it has become evident that in an important subset of patients with TAAD with no recognizable etiology (e.g. hypertension, bicuspid aortic valve) there is frequently a family history of aortopathy. Conceptually, the patients with genetically related TAAD can be divided into syndromic and non-syndromic subgroups. TAAD-related syndromes include Marfan syndrome (FBN1 mutation), Loeys-Dietz syndrome (TGFBR1 and TGFBR2 mutations), Ehlers-Danlos syndrome (COL3A1 mutation) and Turner syndrome (45, X karyotype). Non-syndromic TAADs include mutations of TGFBR2 (that are not associated with Loeys-Dietz syndrome) and mutations of the MYH11, SLC2A10 and ACTA2 genes. In this non-syndromic subgroup, the ACTA2 gene mutation on chromosome 10q22-24 is the most common culprit. 1 The SMC-specific isoform of the α-actin (ACTA2) protein is a major component of the contractile apparatus in vascular SMCs. These SMCs are present throughout the arterial system and are located in the tunica media alongside elastic fibers. ACTA2 heterogeneous mutations interfere with actin filament assembly leading to impaired contraction of the SMCs. Although the mechanism is not completely understood, impaired contraction is thought to lead to vascular wall remodeling, increased wall stiffness and aortic dilation. In the literature, analysis of explanted affected aortic walls has revealed medial degeneration, mild elastolysis, SMC hyperplasia and disarray, and stenotic vasa vasorum due to medial SMC proliferation. In mid-size arteries (cerebral vasculature and coronary arteries), ACTA2 mutations may also be associated with occlusive vascular pathology, which has been attributed to the increased proliferation of SMCs in both the intimal and medial layers of arteries. 2 Clinically, ACTA2 mutations have been associated with diffuse and diverse vasculopathy, including acute aortic syndromes, premature coronary artery disease, transient ischemic attacks and ischemic strokes (including Moyamoya disease), pulmonary hypertension, livedo reticularis and PDA.
Since the original description in 2007, more than 20 different missense mutations of ACTA2 have been described associated with aortopathy. 3 Correlations have emerged between specific ACTA2 mutations and particular vascular involvement. For example, R118Q and R149C mutations predispose primarily to aortic disease and early onset coronary artery disease, whereas mutations altering R258 are associated with aortic disease, PDA, and very early onset cerebrovascular disease, including Moyamoya disease. 4 The R179H ACTA2 mutation, which was found in our patient, was first described in 2010. 4 This specific mutation seems to be associated with a more severe form of vasculopathy compared to other ACTA2 mutations. In these patients, diffuse SMC dysfunction does not only involve the aorta and the cerebrovasculature, but also non-vascular structures that contain SMCs such as the pupils, bladder and gastrointestinal tract. Clinically, these individuals not only develop aortic aneurysms at a very young age, but also manifest congenital mydriasis, hypotonic bladder and congenital gastrointestinal abnormalities.
The case we describe is unusual in that the patient suffered a large limb vessel acute ischemic event. Although it is possible that the patient had a separate unidentified hypercoagulable state, we believe that it was likely related to her ACTA2 mutation. We believe this, as the patient’s hypercoagulable work-up was negative, and mid-size artery vascular disease has been well reported in ACTA2 patients. Up to now, described vascular disease in ACTA2 mutations has been only observed in the cerebrovascular and coronary territories. 5 This is the first report of a vascular limb ischemic event in association with a known ACTA2 mutation. Although the exact mechanism for the patient’s limb ischemia is not clear, it might have been due to embolization from a proximal mural thrombus lining the arterial wall of an ectatic segment. This would possibly represent a different pathophysiology to the cerebrovascular and coronary occlusive disease described in these patients. Another unusual finding in this patient was the ophthalmological abnormalities. As previously mentioned, the patient’s pupillary anomaly has been described in reports of patients harboring this mutation. 4 However, the marked retinal vascular tortuosity findings have not been previously reported in patients with ACTA2 mutations. This may be due to the fact that most of these patients probably did not undergo dedicated fundoscopy as part of their screening. As genetic testing for TAAD becomes more prevalent, the full spectrum of ACTA2 mutations will be better established and it is likely that similar patients will be described.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors have no conflicts of interest to declare.