
Abstract
Objective:
This post hoc analysis of the CHAMPION-NMOSD trial evaluated the safety and efficacy of ravulizumab in patients with aquaporin-4 antibody-positive (AQP4-Ab+) neuromyelitis optica spectrum disorder (NMOSD) previously exposed or naïve to rituximab (RTX).
Methods:
Patients received weight-based intravenous ravulizumab with a loading dose followed by maintenance dosing every 8 weeks. Patients were stratified by prior RTX exposure: no RTX exposure (RTX-naïve) vs RTX exposure > 3 months before initiating ravulizumab (RTX-exposed). Key outcomes included treatment-emergent adverse events (TEAEs), serious TEAEs (TESAEs), relapse rates, and vaccination timing from the last RTX dose.
Results:
Of the 58 patients enrolled, 89.7% were female, with a mean age of 47.4 years, and 21/58 (36.2%) were RTX-exposed. Relapses occurred in 12/21 (57.1%) RTX-exposed patients between their first RTX dose and study entry. The safety profile of ravulizumab was generally similar between RTX-exposed and RTX-naïve groups. Common TEAEs included COVID-19, headache, urinary tract infection, and upper respiratory tract infection. UTIs were more frequent in RTX-exposed individuals. One patient in each group experienced a meningococcal infection. No adjudicated on-trial relapses were reported while on ravulizumab.
Conclusions:
Following initiation of ravulizumab, RTX-exposed and RTX-naïve patients with AQP4-Ab+ NMOSD achieved sustained disease control and demonstrated a manageable safety profile.
Clinical Trial Information:
The CHAMPION-NMOSD Trial; ClinicalTrials.gov identifier: NCT04201262 (registered October 06, 2020).
Keywords
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is a severe autoimmune condition affecting the central nervous system, particularly the optic nerves and spinal cord. The disease is primarily driven by autoantibodies targeting aquaporin-4 water channel proteins expressed on astrocytes, triggering complement activation, astrocyte damage, neuroinflammation, and demyelination. This pathophysiological cascade causes debilitating clinical presentations such as optic neuritis, transverse myelitis, and area postrema syndrome.1 –3
Rituximab (RTX), a chimeric, monoclonal antibody targeting CD20-positive B cells, has been widely used off-label in NMOSD management via pleiotropic mechanisms, in part by depleting autoantibody-producing B cells that activate complement-mediated destruction of astrocytes, oligodendrocytes, and neurons. Although RTX has been examined in small randomized trials such as RIN-1, 4 there has been no registered Phase 3 study to date, and RTX remains unapproved by the United States Food and Drug Administration (FDA) for NMOSD treatment. Despite reducing relapse rates and maintaining established clinical use,5,6 B-cell depleting therapies have important limitations necessitating alternative treatment approaches for certain patient populations. According to FDA-approved labeling, serious, including life-threatening or fatal, bacterial, fungal, and new or reactivated viral infections have been observed during and following completion of treatment with B-cell depleting agents such as rituximab and inebilizumab.7,8 Additional limitations include breakthrough disease activity, 9 infusion-related reactions,10,11 anti-drug antibody formation, 12 and hypogammaglobulinemia.13,14 These safety considerations highlight the clinical need for therapies with alternative mechanisms of action.
Several FDA-approved therapies for NMOSD exist, with varying mechanisms of action and pre-clinical trial patient exposure to RTX. Among approved FDA treatments for NMOSD, CHAMPION-NMOSD (ravulizumab) enrolled the highest proportion of RTX-experienced patients (36.2%); 15 whereas PREVENT (eculizumab [27.1%]),16,17 SakuraStar (satralizumab [13%]), 18 and N-MOmentum (inebilizumab [7.5%])19 –21 enrolled fewer (Table 1). The PREVENT trial demonstrated that prior RTX use did not affect the safety or efficacy of eculizumab.17,22 In the N-MOmentum trial, inebilizumab remained effective in patients exposed to RTX; however, a higher overall infection rate (94%) was reported in these patients compared to those without prior RTX use (70%). 21
Proportion of patients with prior RTX exposure in NMOSD trials.
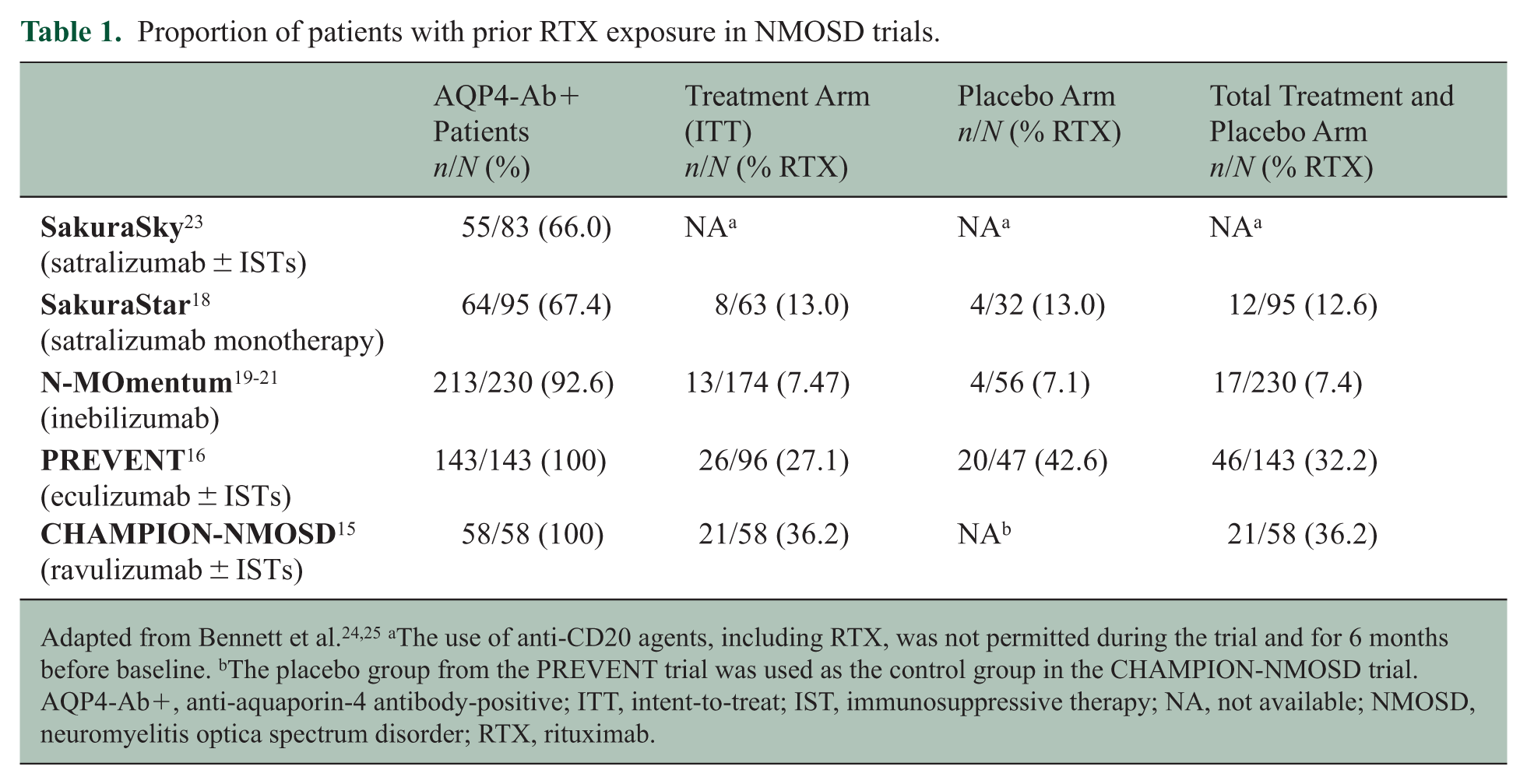
Adapted from Bennett et al.24,25 aThe use of anti-CD20 agents, including RTX, was not permitted during the trial and for 6 months before baseline. bThe placebo group from the PREVENT trial was used as the control group in the CHAMPION-NMOSD trial. AQP4-Ab+, anti-aquaporin-4 antibody-positive; ITT, intent-to-treat; IST, immunosuppressive therapy; NA, not available; NMOSD, neuromyelitis optica spectrum disorder; RTX, rituximab.
Based on the relatively large proportion of patients with prior RTX exposure in the CHAMPION-NMOSD trial, this post hoc analysis aims to evaluate the safety and efficacy of ravulizumab—a long-acting complement C5 inhibitor therapy (C5IT) approved for NMOSD in 2023 (Japan 26 and EU, 27 with US approval in 2024) 28 —in patients with anti-aquaporin-4 antibody-positive (AQP4-Ab+) NMOSD, stratified by prior RTX exposure. Ravulizumab acts as a terminal complement inhibitor by binding to complement protein C5, preventing the formation of the membrane attack complex. In NMOSD, this mechanism is presumed to protect against aquaporin-4 antibody-induced complement-mediated injury.15,28,29 Furthermore, ravulizumab has demonstrated a favorable safety profile relative to eculizumab. 15 This post hoc analysis provides critical insights about clinical outcomes in patients who have prior RTX exposure, and specifically reports on vaccination timing after RTX exposure, relapse prevention, and safety outcomes in this patient population.
Methods
This post hoc analysis of CHAMPION-NMOSD evaluated the safety and efficacy of ravulizumab in patients with AQP4-Ab+ NMOSD who were exposed or naïve to prior RTX treatment, representing the largest dataset to date on RTX-exposed patients with NMOSD initiating complement inhibition or any other FDA-approved therapy for NMOSD. The detailed methods for this study have been described previously for the CHAMPION-NMOSD pivotal trial. 15 Briefly, CHAMPION-NMOSD (NCT04201262) is a Phase 3, multicenter, externally controlled, open-label interventional study assessing the safety and efficacy of ravulizumab in adult patients with AQP4-Ab+ NMOSD. 15 The CHAMPION-NMOSD trial was conducted in accordance with the provisions of the Declaration of Helsinki, the International Conference on Harmonisation guidelines for Good Clinical Practice, and applicable regulatory requirements. The trial was approved by institutional review boards at each participating institution. All patients provided written informed consent before participation. Given the ethical considerations concerning placebo use in NMOSD (i.e. the potential for severe relapses) as well as the availability and efficacy of eculizumab, CHAMPION-NMOSD utilized the placebo arm from the PREVENT trial 16 as an external comparator.
Study design
The CHAMPION-NMOSD trial consisted of four distinct periods, as described in Pittock et al. 15 and shown in Figure 1.
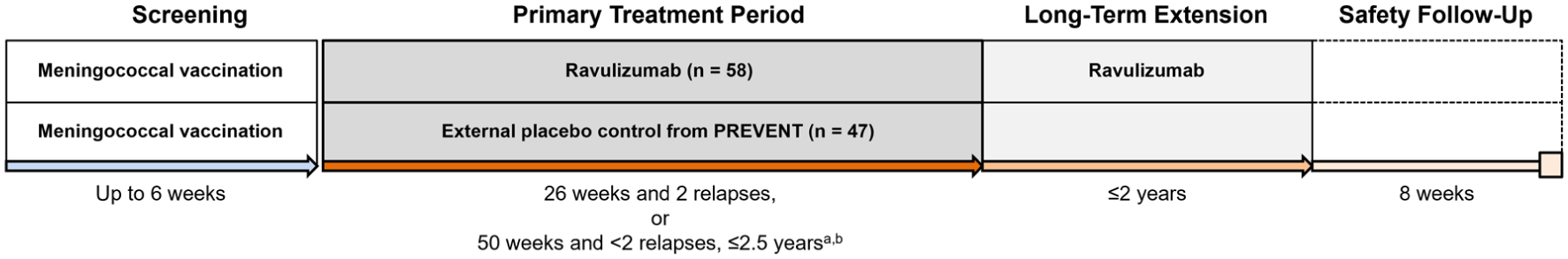
CHAMPION-NMOSD trial design.
All 58 patients who enrolled in CHAMPION-NMOSD received C5IT and were included in this analysis. Data were analyzed through the cutoff date of June 14, 2024. Patients were stratified based on prior RTX exposure. Eligibility criteria for CHAMPION-NMOSD excluded patients with RTX exposure within 3 months prior to screening.
Key safety outcomes included treatment-emergent adverse events (TEAEs), treatment-emergent serious adverse events (TESAEs), and deaths or withdrawals due to TEAEs.
Patient population
Patients eligible for inclusion in the CHAMPION-NMOSD study were adults (⩾ 18 years of age) diagnosed with AQP4-Ab+ NMOSD according to the 2015 international consensus diagnostic criteria. 3 In addition, patients must have experienced ⩾ 1 NMOSD relapse in the previous 12 months.
Patients were stratified into two subgroups based on prior RTX exposure. The RTX-exposed cohort comprised patients who had received RTX more than 3 months prior to screening, as mandated by the CHAMPION-NMOSD study protocol, while the RTX-naïve cohort included patients who had never received RTX. The CHAMPION-NMOSD study protocol 15 required that all patients be vaccinated against Neisseria meningitidis per local vaccination guidelines (e.g. CDC/ACIP in the United States or relevant health authorities in other countries) within 3 years prior to, or at the time of, initiating ravulizumab. Consistent with the PREVENT trial, patients who initiated ravulizumab on the same day as the first meningococcal vaccine or < 2 weeks after the first meningococcal vaccine were to receive appropriate prophylactic antibiotics until 2 weeks after first meningococcal vaccination.
Outcomes
The exploratory endpoints of this post hoc analysis were the safety profile of ravulizumab stratified by those who had and had not received RTX, relapse rates before and after initiating ravulizumab, and vaccination timing in RTX-exposed patients.
Statistical analysis
No hypothesis testing was performed for this post hoc analysis; results are shown descriptively. The safety profile of ravulizumab stratified by those who had and had not received RTX was assessed by summarizing the number and percentage of patients experiencing each TEAE and, in selected cases, by presenting time to first TEAE using Kaplan-Meier curves. The mean (SD) historical relapse rates in the 2 years prior to screening were summarized. Historical relapses, time on study, and supportive immunosuppressive therapies, as well as the timing of meningococcal infections, vaccinations, and duration, were presented in a swimmer plot for the subgroup of RTX-exposed patients.
Results
In total, 58 patients from 36 sites across 11 countries were enrolled in this study and received ravulizumab. Among the 58 patients included in this post hoc analysis from CHAMPION-NMOSD, 21/58 (36.2%) were RTX-exposed; 37/58 (63.8%) were RTX-naïve. The mean (SD) study duration for both groups combined was 167.9 (40.3) weeks, with the RTX-exposed group having a slightly longer duration on study (RTX-exposed, 184.3 [31.4]; RTX-naïve, 158.6 [42.1]). Among patients who were RTX-exposed, the mean (SD) time from last dose of RTX to the first dose of ravulizumab was 7.2 (3.9) months.
Demographics and baseline characteristics
Demographics and clinical characteristics of the study population at baseline are summarized in Table 2. In this analysis, 20/21 (95.2%) RTX-exposed patients and 32/37 (86.5%) RTX-naïve patients were female. The mean (SD) age was 48.2 (13.4) years for RTX-exposed patients and 47.0 (14.3) years for RTX-naïve patients, with 14/21 (66.7%) RTX-exposed patients and 19/37 (51.4%) RTX-naïve patients aged ⩾ 45 years. Twelve of 21 (57.1%) RTX-exposed patients experienced a historical relapse between their first RTX dose and initiation of ravulizumab.
Baseline demographics of RTX-exposed vs RTX-Naïve groups.
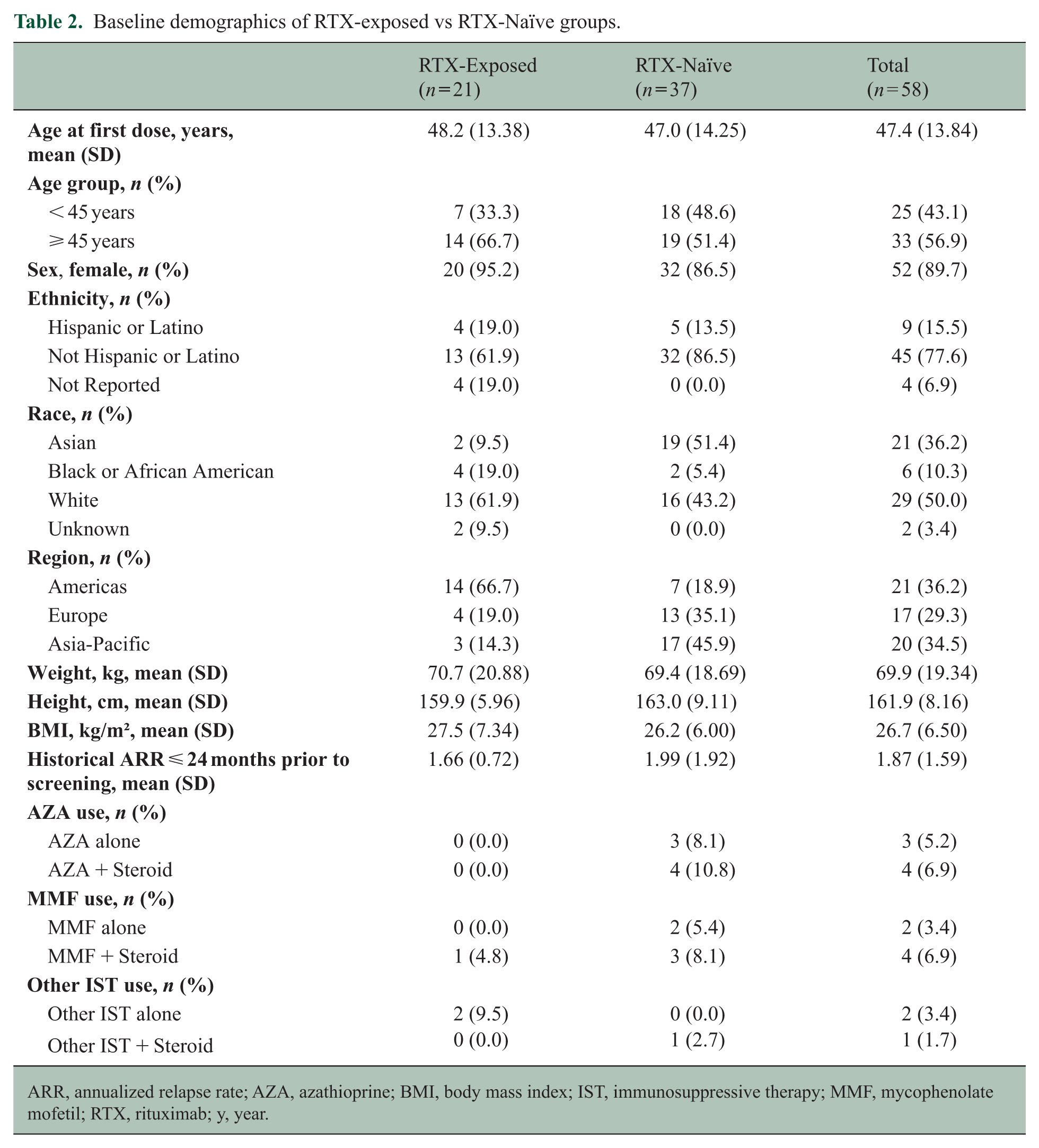
ARR, annualized relapse rate; AZA, azathioprine; BMI, body mass index; IST, immunosuppressive therapy; MMF, mycophenolate mofetil; RTX, rituximab; y, year.
Racial distribution varied between the two groups. Asian patients represented 2/21 (9.5%) RTX-exposed patients compared with 19/37 (51.4%) in the RTX-naïve group. Conversely, 13/21 (61.9%) RTX-exposed patients were White, compared with 16/37 (43.2%) patients in the RTX-naïve group. Black or African American patients comprised 4/21 (19.0%) RTX-exposed patients and 2/37 (5.4%) RTX-naïve patients. Regional differences were also observed, with 14/21 (66.7%) RTX-exposed patients enrolling from the Americas, 4/21 (19.0%) from Europe, and 3/21 (14.3%) from the Asia-Pacific region. An opposite trend was observed for RTX-naïve patients, with only 7/37 (18.9%) enrolling in the Americas, 13/37 (35.1%) in Europe, and 17/37 (45.9%) in Asia-Pacific. Mean (SD) weight, height, and body mass index (BMI) were similar between groups.
The mean (SD) time from initial clinical presentation of NMOSD to first ravulizumab dose was 4.4 (5.3) years in RTX-exposed patients and 5.6 (6.9) years in RTX-naïve patients. In the 24 months prior to screening, the mean (SD) historical annual relapse rate was 1.66 (0.72) and 1.99 (1.92) in RTX-exposed and RTX-naïve patients, respectively.
At baseline, the use of azathioprine (AZA), mycophenolate mofetil (MMF), and other immunosuppressive therapies (IST) was infrequent across both RTX-exposed and RTX-naïve groups. Among RTX-naïve patients, AZA was used alone in 3 (8.1%) patients and in combination with steroids in 4 (10.8%) patients, whereas no AZA use was reported in the RTX-exposed group. MMF monotherapy was received by 2 (5.4%) RTX-naïve patients and by none in the RTX-exposed group; MMF combined with steroids was used by 3 (8.1%) RTX-naïve patients and 1 (4.8%) RTX-exposed patient. Other ISTs were used only rarely: 2 (9.5%) RTX-exposed patients received other IST as monotherapy, and 1 (2.7%) RTX-naïve patient received other IST in combination with steroids. No RTX-naïve patients received other ISTs as monotherapy.
Safety outcomes
A summary of TEAEs and TESAEs, stratified by prior RTX exposure, is provided in Table 3. Most patients in both groups experienced at least one TEAE, of which the majority were mild or moderate in severity and assessed as not related to ravulizumab. In addition, 6/21 (28.6%) RTX-exposed and 9/37 (24.3%) RTX-naïve patients experienced TESAEs, with the majority assessed as not related to ravulizumab. TESAEs were uncommon in both RTX-exposed and RTX-naïve patients, with no apparent clustering within any system organ class (Table 3). In the RTX-exposed cohort, infection and infestations represented the highest proportion of TESAEs (14.3%), and the three individual events occurred in three distinct patients. A similar pattern was observed in the RTX-naïve group, in which infection and infestations occurred in 10.8% of patients, with each event occurring in a different individual. Overall, TESAE rates per 100 patient-years were low and comparable between groups, and no safety trends indicating an increased risk associated with prior RTX exposure were observed.
TEAEs and TESAEs by prior RTX use.
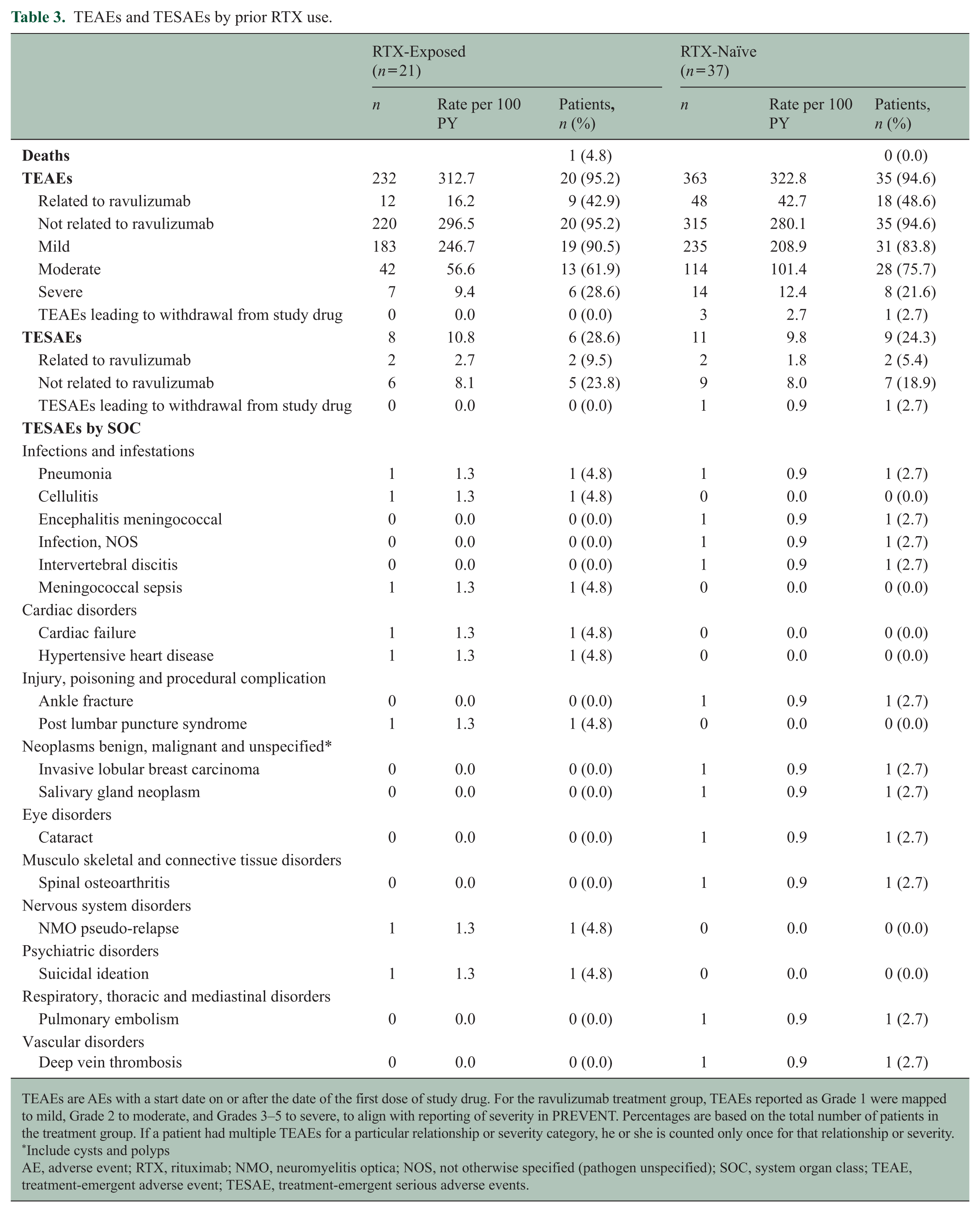
TEAEs are AEs with a start date on or after the date of the first dose of study drug. For the ravulizumab treatment group, TEAEs reported as Grade 1 were mapped to mild, Grade 2 to moderate, and Grades 3–5 to severe, to align with reporting of severity in PREVENT. Percentages are based on the total number of patients in the treatment group. If a patient had multiple TEAEs for a particular relationship or severity category, he or she is counted only once for that relationship or severity.
Include cysts and polyps
AE, adverse event; RTX, rituximab; NMO, neuromyelitis optica; NOS, not otherwise specified (pathogen unspecified); SOC, system organ class; TEAE, treatment-emergent adverse event; TESAE, treatment-emergent serious adverse events.
The proportion of patients with at least one TEAE was similar between groups (RTX-exposed, 20/21 [95.2%]; RTX-naïve 35/37 [94.6%]). The total number of TEAEs was numerically lower in RTX-exposed patients (232 events; 312.7 per 100 PY) than in RTX-naïve patients (363 events; 322.8 per 100 PY). Among TEAEs occurring in ⩾ 10% of all patients (Table 4), COVID-19 was most frequently reported (28/58 [48.3%]), followed by headaches (19/58 [32.8%]) and urinary tract infections (UTI; 10/58 [17.2%]). A numerically higher proportion of RTX-exposed patients reported UTIs compared with RTX-naïve patients (7/21 [33.3%]; 14.8 per 100 PY vs 3/37 [8.1%]; 8.0 per 100 PY); all UTIs were non-serious and Grade 1–2 in severity. In contrast, TEAEs reported more commonly in RTX-naïve vs RTX-exposed patients included COVID-19 (20/37 [54.1%] vs 8/21 [38.1%]) and back pain (7/37 [18.9%] vs 1/21 [4.8%]).
Summary of TEAEs occurring in ⩾ 10% of patients by prior RTX exposure.
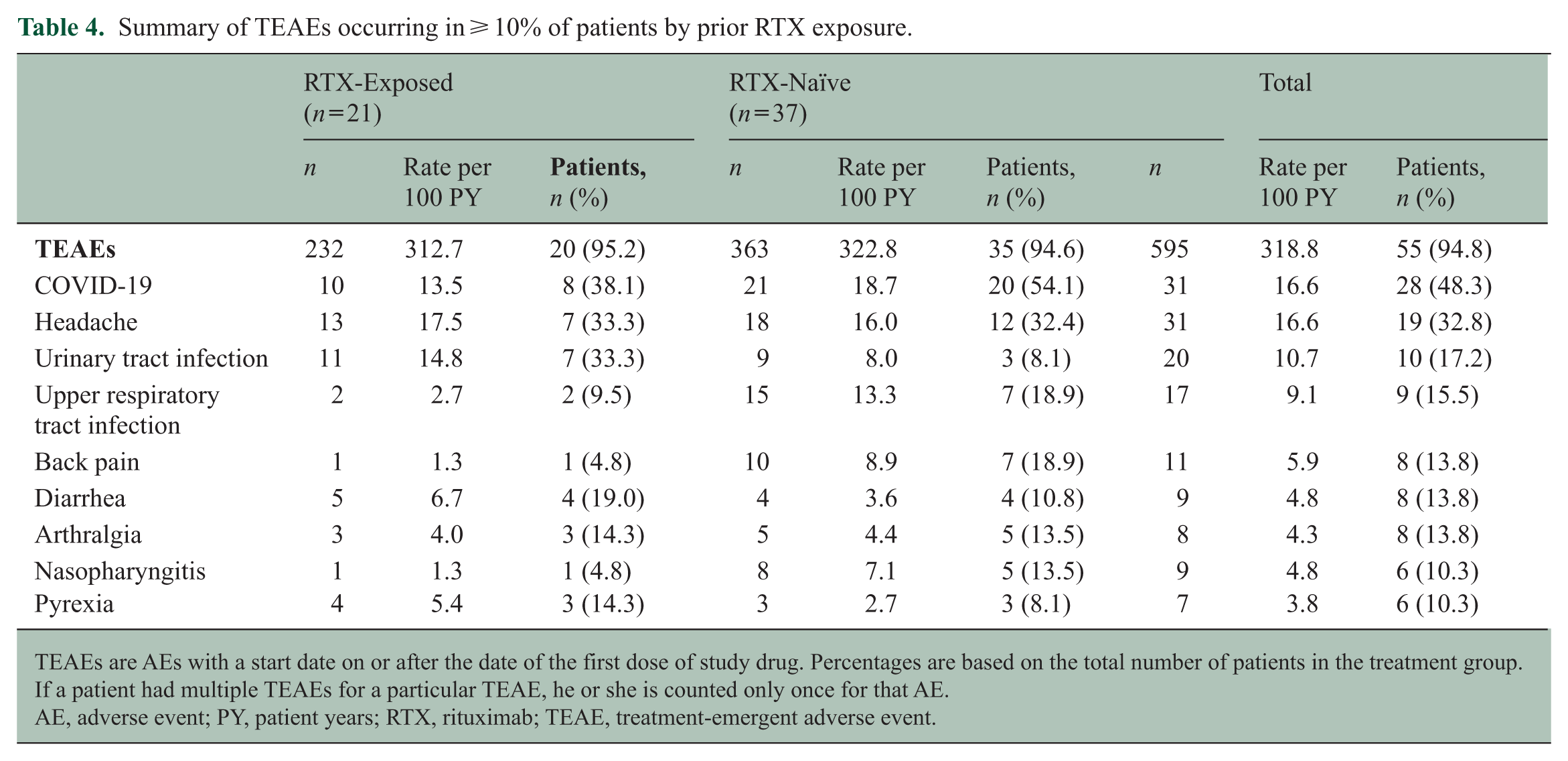
TEAEs are AEs with a start date on or after the date of the first dose of study drug. Percentages are based on the total number of patients in the treatment group. If a patient had multiple TEAEs for a particular TEAE, he or she is counted only once for that AE.
AE, adverse event; PY, patient years; RTX, rituximab; TEAE, treatment-emergent adverse event.
Infections occurring in patients with prior RTX use, stratified by time since last RTX dose, are provided in Table 5. Notably, there were no differences in the rates of COVID-19 infection or UTI between patients who received their last RTX dose at 3–6 months vs 6–12 months before initiating ravulizumab. Moreover, there was no difference in time to first TESAE of infections and infestations system organ class between groups (Figure 2).
TEAEs of infections and infestations occurring in RTX-exposed patients by prior time since last RTX dose.
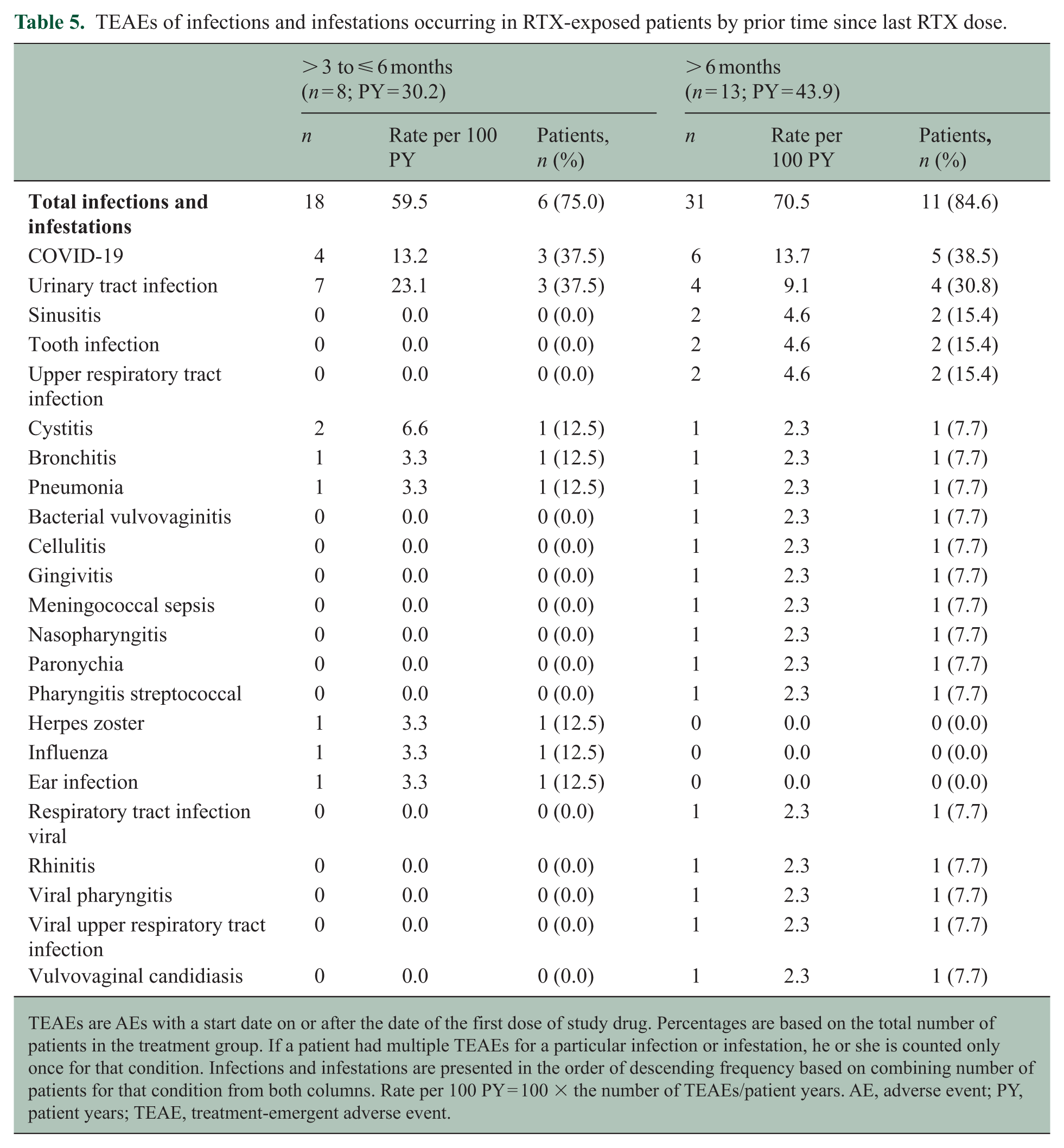
TEAEs are AEs with a start date on or after the date of the first dose of study drug. Percentages are based on the total number of patients in the treatment group. If a patient had multiple TEAEs for a particular infection or infestation, he or she is counted only once for that condition. Infections and infestations are presented in the order of descending frequency based on combining number of patients for that condition from both columns. Rate per 100 PY = 100 × the number of TEAEs/patient years. AE, adverse event; PY, patient years; TEAE, treatment-emergent adverse event.
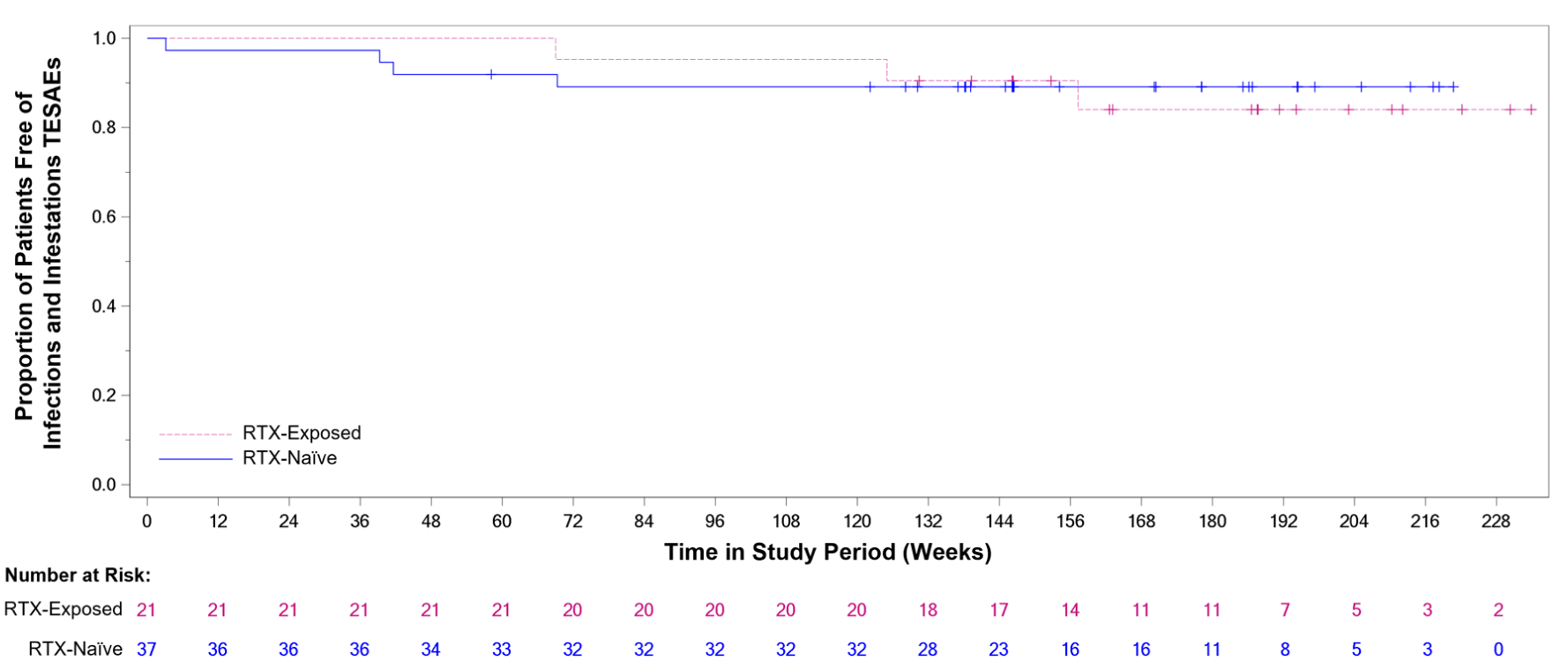
Kaplan-Meier curve for time to first TESAE of infections and infestations in RTX-exposed vs RTX-Naïve patients.
Meningococcal infections are a recognized risk of treatment with complement inhibitors, necessitating vaccination prior to starting treatment. As previously reported in the primary manuscript, 15 two patients developed meningococcal infection with one patient each in the RTX-naïve (Patient A) and RTX-exposed groups (Patient B). Patient A was receiving ravulizumab monotherapy and developed a serotype W135 infection 21 days after their initial ravulizumab dose. Patient B was receiving ravulizumab with concomitant mycophenolate mofetil and prednisolone and developed a serotype B infection 483 days after their first ravulizumab dose. Despite receiving their last RTX dose 13 months prior to initiating ravulizumab, Patient B still had a CD19 B-cell count below the normal range (0.04 × 109/L; normal range: 0.11–0.70 × 109/L) 2 weeks prior to the onset of their meningococcal infection. Both patients were promptly treated with antibiotics and intensive care, recovering fully without sequelae. Patient A withdrew from the study following recovery, while Patient B elected to continue receiving ravulizumab in the study.
Meningococcal vaccination timing and antibiotic prophylaxis in RTX-exposed patients
Vaccination timing for all 21 patients with prior RTX exposure is included in Figure 3, with vaccination timing characteristics captured for 19 patients vaccinated for the first time after RTX exposure. Among the 19 remaining patients previously exposed to RTX, the mean (SD) time from last RTX dose to first subsequent meningococcal vaccination was 5.58 (3.78) months, with a range of 2.7–19.9 months. Most patients (13/19 [68.4%]) received their first meningococcal vaccination within 6 months of their last RTX dose. More specifically, 3/19 (15.8%) patients received their first post-RTX vaccination within 3 months and 10/19 (52.6%) between 3 and 6 months; 6/19 (31.6%) patients received their first vaccination more than 6 months after their last RTX dose. Furthermore, three patients received oral antibiotic prophylaxis for meningococcal prevention during the study: one RTX-exposed patient (Patient B, above) received phenoxymethylpenicillin potassium (500 mg BID for 10 months), then transitioned to amoxicillin (250 mg QD, ongoing) following recovery from meningococcal infection, and two RTX-naïve patients received short-course prophylaxis. Of the two RTX-naïve patients, one received ciprofloxacin (500 mg QD for < 1 month) and one received azithromycin (500 mg QD for 3 days).
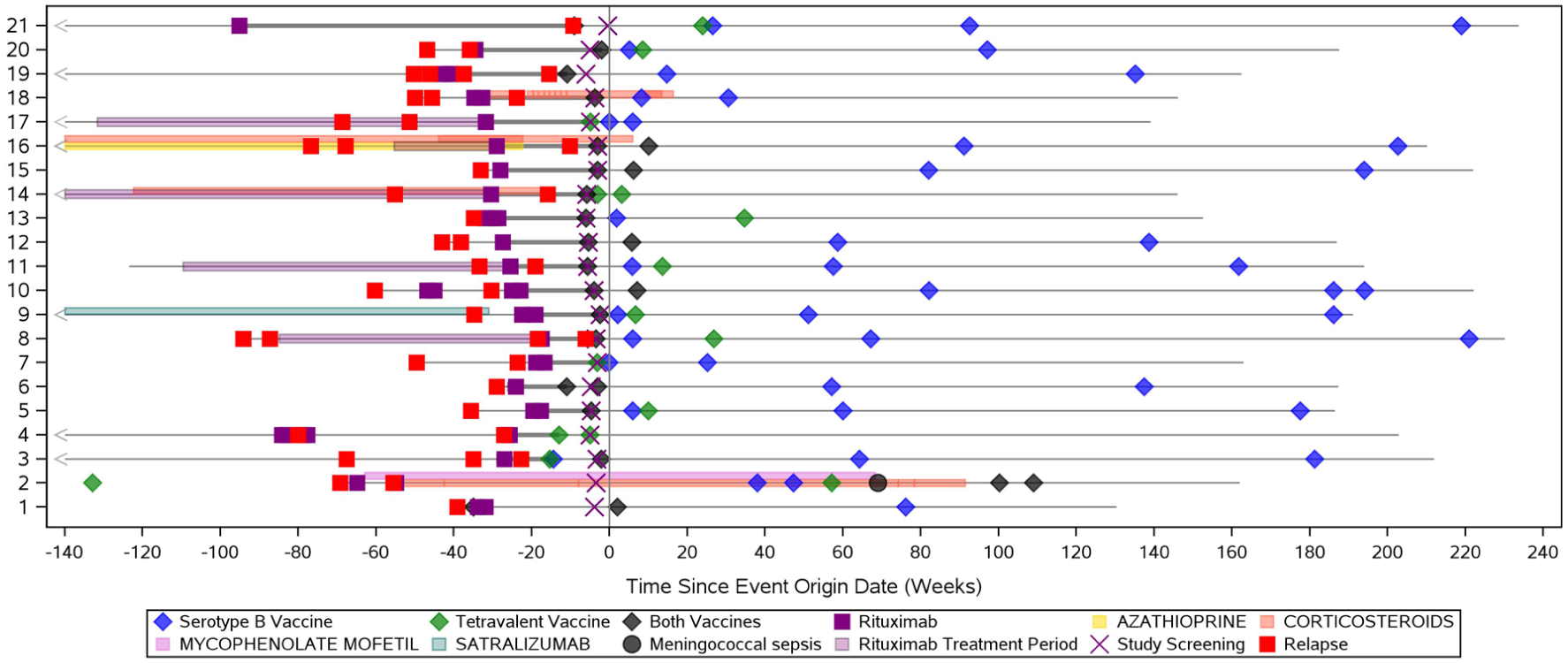
Patient-level visualization of meningococcal vaccine administration, immunosuppressive therapy, and relapse history in RTX-exposed patients.
Efficacy outcomes
In CHAMPION-NMOSD, there were no positively adjudicated physician-determined on-trial relapses in any patient, regardless of prior RTX exposure. 15
Discussion
This post hoc analysis represents the largest descriptive subgroup analysis of safety and efficacy in patients with AQP4-Ab+ NMOSD previously treated with RTX before initiating C5IT or other FDA-approved NMOSD therapies. CHAMPION-NMOSD included 21 RTX-exposed patients, exceeding prior RTX-exposed samples reported in trials of satralizumab (n = 8) and inebilizumab (n = 13) and thus providing the most extensive clinical trial evidence to date evaluating an FDA-approved therapy for NMOSD in patients with prior RTX exposure. Findings were directionally consistent with prior reports of complement inhibition in NMOSD, with broadly comparable outcomes between RTX-exposed and RTX-naïve patients.15,21,30 No increase in overall TEAE incidence among RTX-exposed patients following initiation of ravulizumab was observed, although a higher proportion of UTIs was reported in RTX-exposed patients; these observations were descriptive from a small post hoc subgroup without formal hypothesis testing. These results support consideration of ravulizumab in patients with AQP4-Ab+ NMOSD who have previously received RTX, further validating the role of complement inhibition in the management of NMOSD.30,31
For RTX-exposed patients with ongoing disease activity or non-relapse–related complications such as infections or hospitalizations, ravulizumab, an FDA-approved treatment, may be considered as part of treatment sequencing.32 –34 The increased frequency of UTIs observed in RTX-exposed patients aligns with previous literature identifying infections—commonly UTIs and respiratory infections—during or following RTX treatment.33,34 This consistency underscores the importance of close monitoring and early management where indicated.
The data reported in this analysis align with expert recommendations to switch to a medication with a different mechanism of action if patients experience treatment failure (i.e. disease activity) or side effects.35,36 Beyond the immediate findings of this analysis, it is important to consider the underlying pathophysiology of NMOSD when selecting treatment strategies. Complement activation plays a central role in NMOSD pathophysiology by driving astrocyte damage, which can result in clinical disability. Targeted complement inhibition can minimize the risk of NMOSD attacks, thereby preventing astrocyte loss and subsequent neuronal death. 37
Patients treated with RTX may experience prolonged B-cell depletion for several months after their previous dose. 38 Although lymphocyte subsets were not collected in the CHAMPION-NMOSD trial, it is possible that RTX-exposed patients may have been at various stages of B-cell reconstitution at the time of vaccination, potentially influencing vaccine responsiveness. The VELOCE study 39 shows that patients with relapsing multiple sclerosis treated with the CD20-targeting agent ocrelizumab mounted attenuated, but potentially protective, humoral responses to clinically relevant non-live vaccines, including tetanus toxoid, pneumococcal polysaccharide, and seasonal influenza vaccines, despite confirmed peripheral B-cell depletion. These findings indicate that B-cell depletion does not necessarily preclude meaningful vaccine responses. Moreover, the potential for protective immunity may also be supported through T-cell-mediated mechanisms, particularly for protein-based and conjugate vaccines such as the meningococcal conjugate vaccine, which are known to elicit immunologic memory independent of B-cell counts.40 –42 For Patient B described previously, the first series of serogroup B vaccinations was administered during a period of reduced CD19 B cells, 6–8 months prior to the meningococcal serogroup B episode. It has been postulated that the patient’s meningococcal vaccination response was further compromised by concurrent therapy with mycophenolate, which impairs B- and T-cell proliferation and may have contributed to prolonged B-cell depletion after RTX, given the B-cell reconstitution observed after mycophenolate discontinuation. 43 Importantly, in clinical practice, for patients with NMOSD who require urgent initiation of ravulizumab, risk mitigation strategies such as meningococcal vaccination and prophylactic antibiotics are permissible per the US Prescribing Information.28,44 Post hoc analyses and clinical trial data have demonstrated regional variability in provider-driven prophylactic antibiotic practices, including differences in antibiotic agents selected and the duration of exposure. 45 Taken together, these insights suggest that vaccination prior to treatment with ravulizumab in patients with NMOSD has the potential for protective immunity even in the context of prior RTX exposure.
This retrospective analysis introduces certain inherent limitations, including small sample size, heterogeneity in RTX washout intervals, and potential confounding from prior and concomitant therapies. The study was not powered for between-group comparisons and results should be interpreted descriptively. Despite these constraints, this remains the largest controlled-trial cohort of RTX-exposed patients initiating ravulizumab.
Future work should include observational studies and real-world data analyses to characterize long-term outcomes after treatment transitions, as well as biomarker-based investigations of immune reconstitution to refine sequencing strategies and timing between RTX and complement inhibition.
The results of this post hoc analysis support consideration of ravulizumab for patients previously treated with RTX and may inform future research aimed at advancing personalized treatment strategies for patients with NMOSD.
Footnotes
Acknowledgements
Medical writing support was provided by Alexander Schwartz, Ph.D., and Cassandra Uchida, Ph.D., of Sixsense Strategy Group, Inc., a Herspiegel company, and funded by Alexion, AstraZeneca Rare Disease.
Author Contributions
The authors contributed to the conception or design of the work or the acquisition, analysis, or interpretation of data for the work; drafting the work or reviewing it critically for important intellectual content; and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JLB has received personal compensation for serving as a consultant for Alexion, AstraZeneca Rare Disease, Amgen, Beigene, Reistone Bio, Chugai, Genentech, ImCyse, ImmPACT Bio, Mitsubishi Tanabe, Novartis, TG, Antigenomycs, and Roche; serving on a Scientific advisory or data safety monitoring board for CorEvitas, MIAC, Clene, and Roche; serving on a Speakers Bureau for Alexion, AstraZeneca Rare Disease, Efficient LLC, Touch IME, and Vindico; serving as an expert witness for Marie Bush, Pavich Law, Marks Gray, and Knight, Nicastro, MacKay; received institutional research support from Novartis, Mallinckrodt, Genentech, Alexion, AstraZeneca Rare Disease, National Institutes of Health, Department of Defense, and the National Multiple Sclerosis Society; received intellectual property interests from a discovery or technology relating to health care; and received publishing royalties from a publication relating to health care. SB has received research support from Alexion, AstraZeneca Rare Disease, NIH, Roche, UCB, and NeuroLambda Therapeutics; publishing honoraria from American Academy of Neurology and UpToDate. JDS has received consulting fees from Biogen, Teva, BMS/Celgene, Roche, Novartis, Janssen, Merck, Alexion, UCB, Sanofi-Genzyme, Horizon Therapeutics; and honoraria from Biogen, Teva, BMS/Celgene, Roche, Novartis, Janssen, Merck, and Alexion, AstraZeneca Rare Disease. ML has received fees from Alexion, AstraZeneca Rare Disease; Genentech, Inc.; and Horizon Therapeutics. SJP has received personal compensation for serving as a consultant for F. Hoffman-LaRoche AG, Genentech, Sage Therapeutics, Astellas and Arialis. He’s received personal compensation for serving on scientific advisory boards or data safety monitoring boards for F. Hoffman-LaRoche AG, Genentech, and UCB. His institution has received compensation for serving as a consultant for Astellas, Alexion/Astra Zeneca Rare Diseases, and Horizon/Amgen. All compensation is paid to Mayo Clinic. He has received research support from Alexion/Astra Zeneca Rare Diseases, Horizon/Amgen, F. Hoffman-LaRoche AG, Genentech and Adimmune. He has a patent, Patent# 8,889,102 (Application#12-678350, Neuromyelitis Optica Autoantibodies as a Marker for Neoplasia)—issued; a patent, Patent# 9,891,219B2 (Application#12-573942, Methods for Treating Neuromyelitis Optica [NMO] by Administration of Eculizumab to an individual that is Aquaporin-4 [AQP4]-IgG Autoantibody positive)-issued and from which he has received royalties. AZ has received research funding from Alexion, AstraZeneca Rare Disease, Biogen, Chugai, Genentech, Medimmune, NIH, and PCORI, and honoraria for speaker bureau or advisory board from Biogen, Celgene/Bristol Myers Squibb, Genentech-Roche, Merck-Serono, Novartis, Teva Pharma, Sanofi-Genzyme, Viela Bio/Horizon/Amgen, and TG therapeutics. UA, MJT, and BP are employees of Alexion, AstraZeneca Rare Disease, and may hold stock or stock options in AstraZeneca. KA was an employee of Alexion, AstraZeneca Rare Disease at the time of research and part of manuscript development and may hold stock or stock options in AstraZeneca.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Alexion, AstraZeneca Rare Disease. Alexion, AstraZeneca Rare Disease was involved in the study design; collection, analysis and interpretation of data; writing, reviewing, and approving the publication; and decision to submit the article for publication.
Ethical Considerations
The CHAMPION-NMOSD trial was conducted in accordance with the provisions of the Declaration of Helsinki, the International Conference on Harmonisation guidelines for Good Clinical Practice, and applicable regulatory requirements. The trial was approved by institutional review boards at each participating institution.
Consent to Participate
All patients provided written informed consent before participation.
Consent for Publication
Not applicable.