
Abstract
Background:
Following natalizumab failure, it is unknown whether switching to alternative high-efficacy therapies offers superior effectiveness over continuing natalizumab.
Objective:
To compare different treatment strategies following natalizumab failure.
Methods:
Patients suffering a relapse during natalizumab treatment with adequate follow-up were identified from the MSBase registry. Following natalizumab failure, natalizumab continuation was compared to switching to anti-CD20 therapies/alemtuzumab/lower-efficacy therapies and treatment discontinuation. The primary outcome was the risk of further relapses. Secondary outcomes included risk of subsequent magnetic resonance imaging (MRI) activity, confirmed disability worsening and disease-activity-free survival. Multivariable proportional hazards models compared outcomes during time-varying therapy exposures. Four sensitivity analyses were conducted with varied inclusion criteria and treatment failure definitions.
Results:
Of 1553 patients experiencing a relapse during natalizumab treatment, 1037 met the inclusion criteria. Following natalizumab failure, switch to anti-CD20 therapy was associated with a lower relapse risk (heart rate (HR) = 0.48, 95% confidence interval (CI) = 0.27–0.84) compared to continuing natalizumab; no differences were observed in MRI or disability outcomes. Treatment de-escalation or cessation was associated with increased relapse risk (HR = 1.46, 95% CI = 1.15–1.85; HR = 2.08, 95% CI = 1.22–3.55, respectively). We did not find evidence of a difference for switching to alemtuzumab. Sensitivity analyses replicated primary findings.
Conclusion:
This exploratory study indicates that switching to anti-CD20 therapies following natalizumab failure is associated with a >50% reduction in relapse risk. No differences were seen in secondary outcomes, despite consistent trends. Clinicians may consider anti-CD20 therapies following natalizumab failure, noting further research is needed to confirm differences in MRI and disability outcomes.
Introduction
Multiple sclerosis (MS) is a demyelinating disease of the central nervous system characterised by distinct episodic deteriorations (relapses) and gradual progression of neurological disability. Disease-modifying treatments (DMTs) have shown varying efficacy in suppressing relapses and preventing disability accumulation.
Among currently available DMTs, natalizumab, alemtuzumab, anti-CD20 therapies (ocrelizumab, ofatumumab and rituximab) and autologous haematopoietic stem cell transplantation have shown the highest efficacy in preventing both relapses and disability accumulation. 1 While these high-efficacy therapies have never been compared in a randomised controlled trial, emulated trials using observational data have demonstrated similar mean effectiveness of natalizumab and alemtuzumab, 2 and natalizumab and ocrelizumab.3 –6 Selecting between high-efficacy DMTs is therefore largely based on risk factor profile and individual patient characteristics (e.g. John Cunningham virus (JCV) serostatus, infusion convenience). 7
Suboptimal disease control on lower-efficacy DMTs can be managed with a switch to more potent DMTs.8 –12 However, there is little evidence to guide decision-making once patients suffer new disease activity while already treated with high-efficacy DMTs.
Autologous haematopoietic stem cell transplantation (AHSCT) is the only treatment demonstrating marginally higher efficacy 13 and therefore presents the only escalation strategy when high-efficacy DMTs fail. AHSCT is, however, associated with significantly increased risks and is only accessible in highly specialised centres. 13 Whether switching between high-efficacy DMTs to a treatment with a different mechanism of action provides any benefits to patients who fail any of these treatments remains unknown.
We therefore conducted this study to evaluate different treatment approaches following failure of high-efficacy DMTs to control MS relapses. Given the scarcity of switches from anti-CD20 therapy to natalizumab (partly due to concerns regarding the validity of JCV serology in this circumstance),14,15 we focused on therapeutic decisions following failure of natalizumab. We hypothesised that, following relapses during natalizumab treatment, switching to a different high-efficacy DMT would lead to improved clinical control of relapsing MS. As a reference, we compared continued treatment with natalizumab to switching to anti-CD20 therapy, alemtuzumab, or lower-efficacy DMTs.
Methods
Ethics statement
The MSBase registry 16 (registered with WHO ICTRP, ID ACTRN12605000455662) was approved by both the Melbourne Health Research Ethics Committee and local ethics committees in participating centres (as per local regulations). Enrolled patients provided written informed consent.
Patient population and data collection
Longitudinal data from 84,324 patients from 157 MS centres in 45 countries were extracted from MSBase in February 2023. Data quality procedures were applied as described previously (Supplemental Table S2). 17
All data were recorded as part of routine clinical practice, with most centres practising real-time data entry. MSBase protocol stipulates minimum annual updates of the dataset, but patients with less frequent visits were not excluded. The data entry portal was the iMed/MSBase data entry system. Only data recorded prospectively from the date of first clinical visit were included, except for the date of first MS symptom. Disability was scored by accredited scorers (online Neurostatus certification required) using the Expanded Disability Status Scale (EDSS).
Inclusion criteria
The minimum dataset consisted of birthdate, sex, date of first MS symptom, and treatment and relapse information. Relapse-onset patients who received natalizumab were identified in the MSBase cohort. Natalizumab failure was defined as a relapse occurring from 90 days after starting natalizumab up to 30 days after the final dose, 18 though an alternate definition including radiological activity was utilised in a sensitivity analysis (detailed below). All included patients suffered a relapse, defined as the occurrence of new symptoms or exacerbation of existing symptoms persisting for ⩾24 hours in the absence of concurrent illness or fever and occurring ⩾30 days after a previous relapse, 19 while treated with natalizumab. The first such relapse was identified as the study baseline. Only relapses after January 2006 were included due to the limited availability of alternative high-efficacy therapies prior to this date. Included patients required at least three in-person disability assessments: a baseline EDSS score (not preceded by a relapse within 30 days) in the year before baseline, and at least two subsequent EDSS scores ⩾6 months apart. The EDSS score nearest the event was used as the baseline EDSS.
Patients were excluded if previously enrolled in a randomised controlled trial, had previously received AHSCT or alemtuzumab, or received mitoxantrone treatment within 3 years of index event.
Study outcomes
The primary study outcome was the occurrence of subsequent relapses following the index relapse on natalizumab. Exploratory secondary outcomes were (a) the occurrence of new disease activity on magnetic resonance imaging (MRI), defined as the presence of ⩾3 new20 –22 or ⩾1 gadolinium-enhancing lesion(s) on MRI brain at a single timepoint; (b) sustained EDSS worsening, defined as an increase in EDSS ( ⩾1.5 point increase if the last recorded EDSS was 0; ⩾1 point increase if EDSS 1–5.5; and ⩾0.5 points if EDSS ⩾6) confirmed over ⩾6 months (identified with the MSoutcomes package for R);23,24 and (c) disease-activity-free survival (relapse, MRI activity or sustained EDSS worsening). MRI scans were performed and reported in keeping with local protocols, using 1.5T–3T scanners, including T1- and T2-based sequences. For disease-activity-free survival, the first MRI and EDSS score following the baseline relapse were discarded.
Patients were grouped by therapeutic decision following natalizumab failure: continuing natalizumab, switching to anti-CD20 therapy (ocrelizumab or rituximab; no switches to ofatumumab were recorded), switching to alemtuzumab or de-escalation to lower-efficacy DMTs (moderate- and low-efficacy DMTs; defined below). Patients were considered untreated in the period between natalizumab discontinuation and commencing subsequent therapy. Only treatment switches occurring within 8 months of natalizumab failure were included; this was based on the presumed duration of natalizumab treatment effect of 2 months from last dose,25,26 and to allow up to a 6-month gap between the end of natalizumab effect and commencement of next therapy.
Patient characteristics
Information about age, EDSS, disease duration at baseline, preceding DMT use and sex was obtained. Individual pre-baseline annualised relapse rates were calculated during the 6 months prior to natalizumab failure. Treatment use immediately preceding natalizumab was stratified into high-efficacy DMTs (anti-CD20 therapy, natalizumab, alemtuzumab, mitoxantrone), moderate-efficacy DMTs (cladribine, fingolimod, dimethyl fumarate) and low-efficacy DMTs (interferon beta, glatiramer acetate, teriflunomide).
Statistical analysis
Statistical analysis was carried out using R (3.1.0, http://www.R-project.org). All hypotheses were tested at the two-tailed 0.05 level of statistical significance.
The associations of patient characteristics and treatment decision following natalizumab failure with the hazard of the study outcomes were analysed using multivariable conditional proportional hazards models. Cox proportional hazards models were used for the secondary outcome of disease-activity-free survival. Data were grouped by treatment states after natalizumab failure (natalizumab continuation/anti-CD20 therapy/alemtuzumab/low-efficacy therapy/untreated) and treated as time-varying exposures, allowing patients to switch between treatment groups during follow-up (Figure 1). Patients were censored at the earliest of either a further treatment change after the initial post-relapse treatment decision, or the last recorded EDSS score (per-protocol contrast of interest). The following baseline predictors were included: sex, age and disease duration at time of natalizumab failure, baseline EDSS score, annualised relapse rate pre-natalizumab failure, number of distinct DMTs utilised before natalizumab and efficacy of the latest treatment prior to natalizumab. Models were adjusted for the study centre as a frailty term.
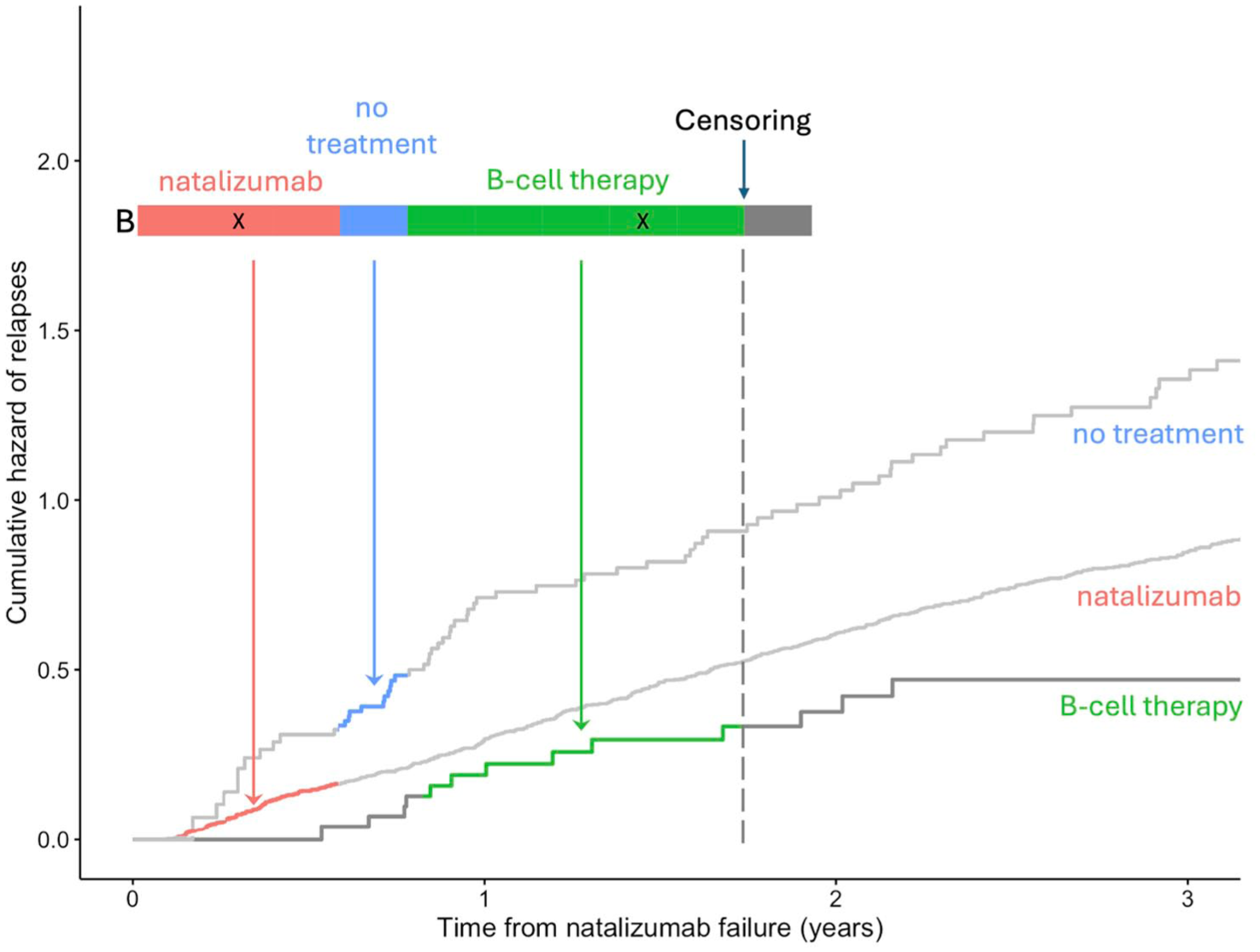
Visualisation of the study design.
Four exploratory sensitivity analyses were undertaken for the primary outcome with varied inclusion criteria or treatment failure definition: (1) only including patients treated with natalizumab for ⩾12 months prior to index relapse; (2) only including patients without any preceding high-efficacy DMT use; (3) defining natalizumab failure as either a relapse or the occurrence of new MRI activity; and (4) defining relapses as only those requiring treatment or clinician-rated as moderate or severe.
Results
Patients
Of 8512 patients ever treated with natalizumab in the MSBase cohort, 1553 patients (18.2%) experienced relapses during treatment with natalizumab. A total of 2551 relapses were recorded across 28,276 natalizumab patient-years, equating to 0.09 relapses/patient-year. Median time to first relapse on natalizumab was 1.04 years (quartiles: 0.57–2.02). A total of 1037 patients fulfilled the inclusion criteria (Figure 2). Table 1 summarises demographic and clinical data for the study cohort. Supplemental Table S3 summarises the characteristics of the 516 excluded patients who relapsed on natalizumab (largely similar to the included cohort).
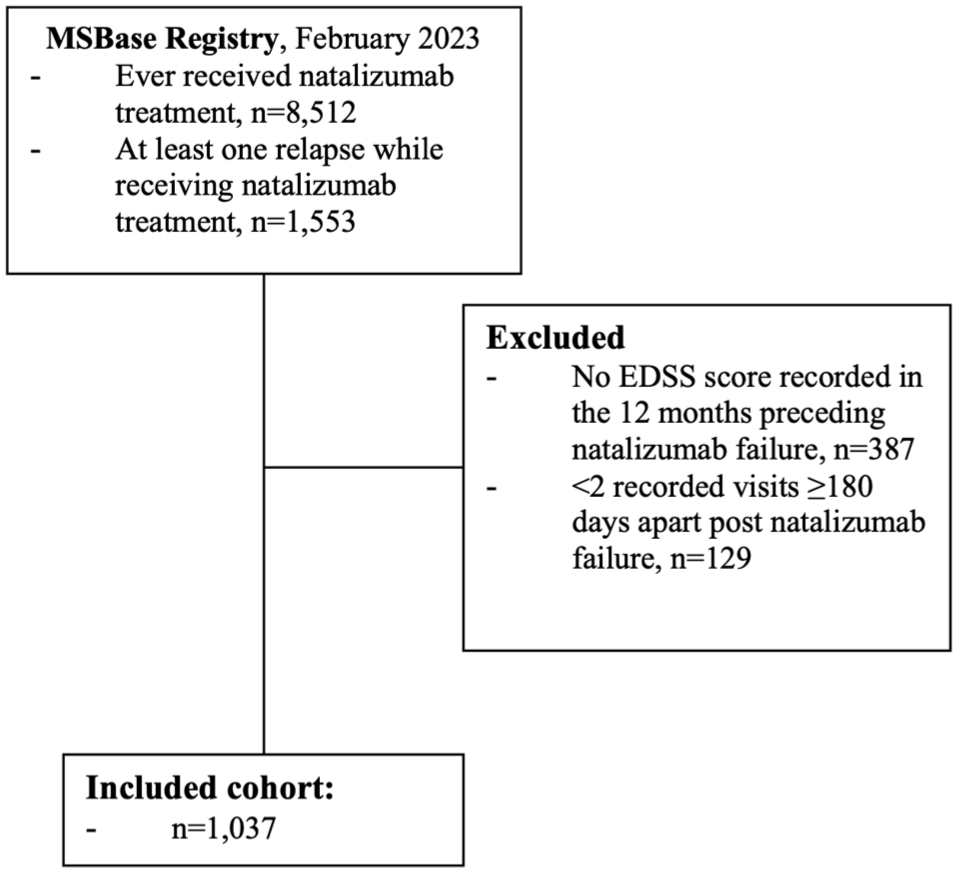
CONSORT diagram of patient disposition.
Characteristics of the Study Population.
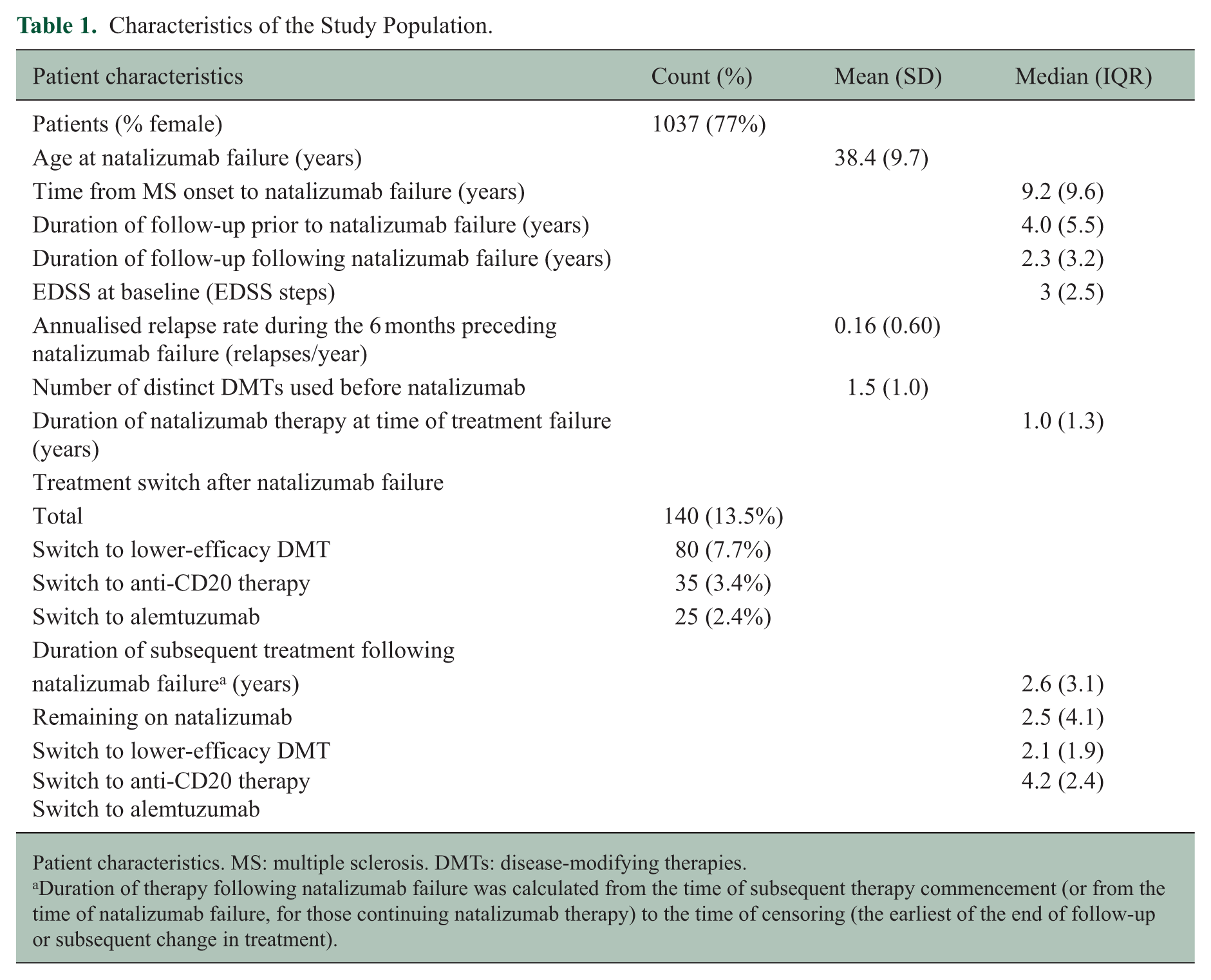
Patient characteristics. MS: multiple sclerosis. DMTs: disease-modifying therapies.
Duration of therapy following natalizumab failure was calculated from the time of subsequent therapy commencement (or from the time of natalizumab failure, for those continuing natalizumab therapy) to the time of censoring (the earliest of the end of follow-up or subsequent change in treatment).
Treatment decisions post-natalizumab failure
Most patients (782) continued natalizumab therapy for a minimum of 8 months following treatment failure. One hundred and five patients ceased natalizumab and did not commence a new treatment within the treatment switch window. Of 140 patients commencing a new treatment, 35 switched to anti-CD20 therapy (24 ocrelizumab, 11 rituximab) and 25 switched to alemtuzumab; 80 de-escalated to a lower-efficacy therapy. The remainder switched to mitoxantrone, underwent AHSCT, or enrolled in a randomised controlled trial, and were thus censored at this time. Supplemental Table S4 compares the characteristics of patients switching therapies and remaining on natalizumab.
Primary outcome: cumulative hazard of relapses
Results of the primary multivariable model are displayed in Table 2. A total of 805 relapses were recorded (detailed event numbers for all analyses are provided in Supplemental Table S5). Following natalizumab treatment failure, switching to anti-CD20 therapy was associated with a reduced risk of relapses (heart rate (HR) = 0.48, 95% confidence interval (CI) = 0.27–0.84) compared to remaining on natalizumab (Figure 3). There was no evidence for a difference in relapse risk following the switch to alemtuzumab. Treatment de-escalation and periods off treatment were associated with increased relapse risk (HR = 1.46, 95%CI = 1.15–1.85; HR = 2.08, 95%CI = 1.22–3.55, respectively) compared to continuing natalizumab. The effect of rituximab and ocrelizumab was near-identical when analysed separately in a post hoc sensitivity analysis (Supplemental Table S6).
Determinants of Relapse Hazard After Failure of Natalizumab.
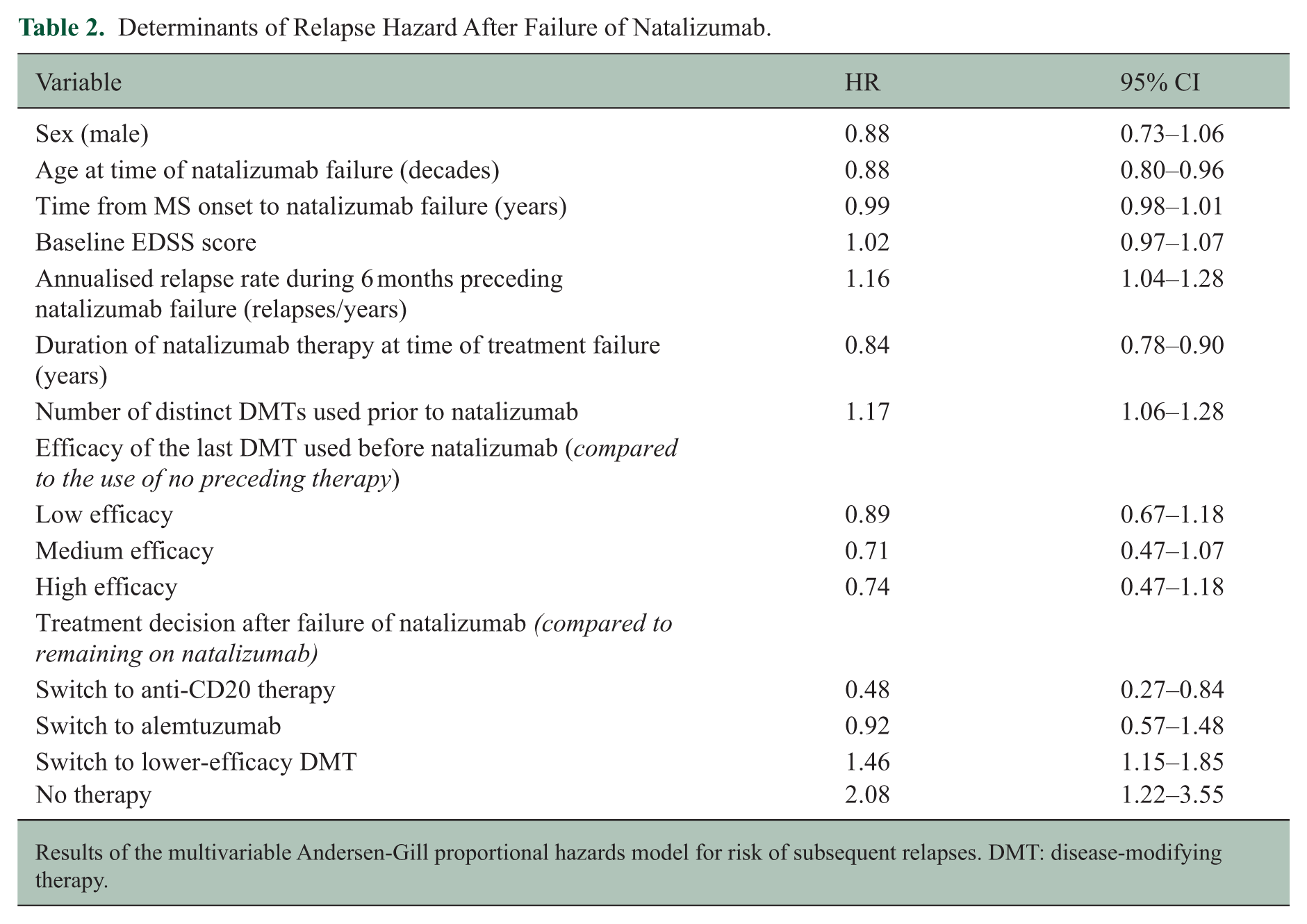
Results of the multivariable Andersen-Gill proportional hazards model for risk of subsequent relapses. DMT: disease-modifying therapy.
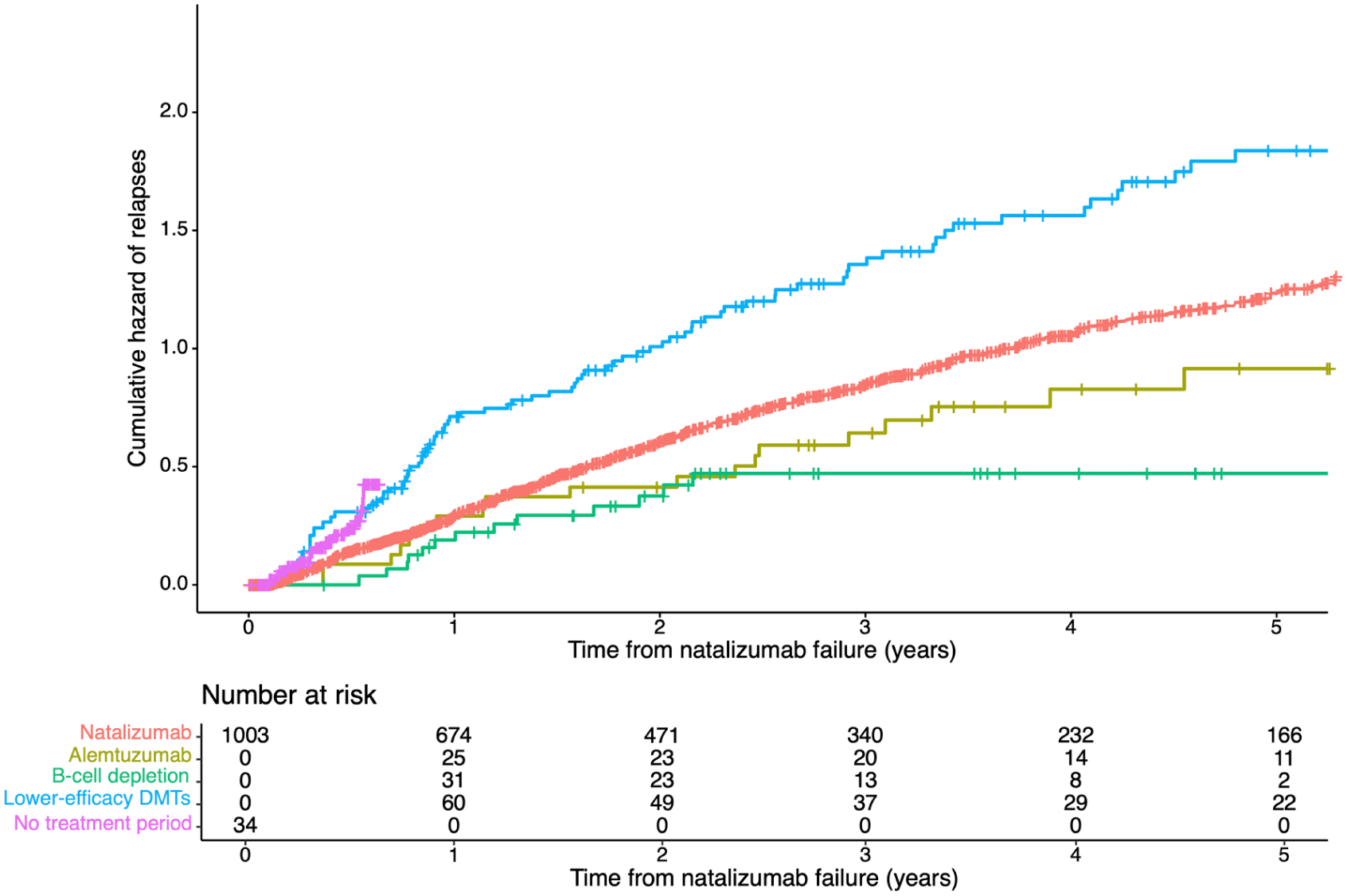
Cumulative hazard of relapses.
Higher relapse rate prior to natalizumab failure, and a higher number of preceding therapies, were associated with increased relapse risk (HR = 1.16, 95% CI = 1.04–1.28; HR = 1.17, 95% CI = 1.06–1.28, respectively). The risk of relapses was lower in older patients (HR = 0.88, 95% CI = 0.80–0.96 per decade) and patients with a longer duration of natalizumab treatment prior to its failure (HR = 0.84, 95% CI = 0.78–0.90 per year).
Secondary/exploratory outcomes
Radiological disease activity
Only 809 patients had at least one post-baseline MRI recorded and were included in this analysis. Results are displayed in Table 3. There were 172 instances of new disease activity on MRI captured during follow-up (Supplemental Table S5). We found no evidence for an association between the switch to anti-CD20 therapies and the risk of MRI activity, though the coefficients were consistent with the associations identified by the primary analysis (HR = 0.76, 95% CI = 0.23–2.51). Treatment de-escalation and cessation were associated with an increased risk of new disease activity on MRI (HR = 2.05, 95% CI = 1.13–3.72; HR = 3.32, 95% CI = 1.59–6.91). Longer disease duration at the time of natalizumab failure and fewer preceding treatments were associated with a reduced risk of new MRI activity.
Determinants of Hazard of Radiological Disease Activity and Sustained EDSS Worsening After Failure of Natalizumab.
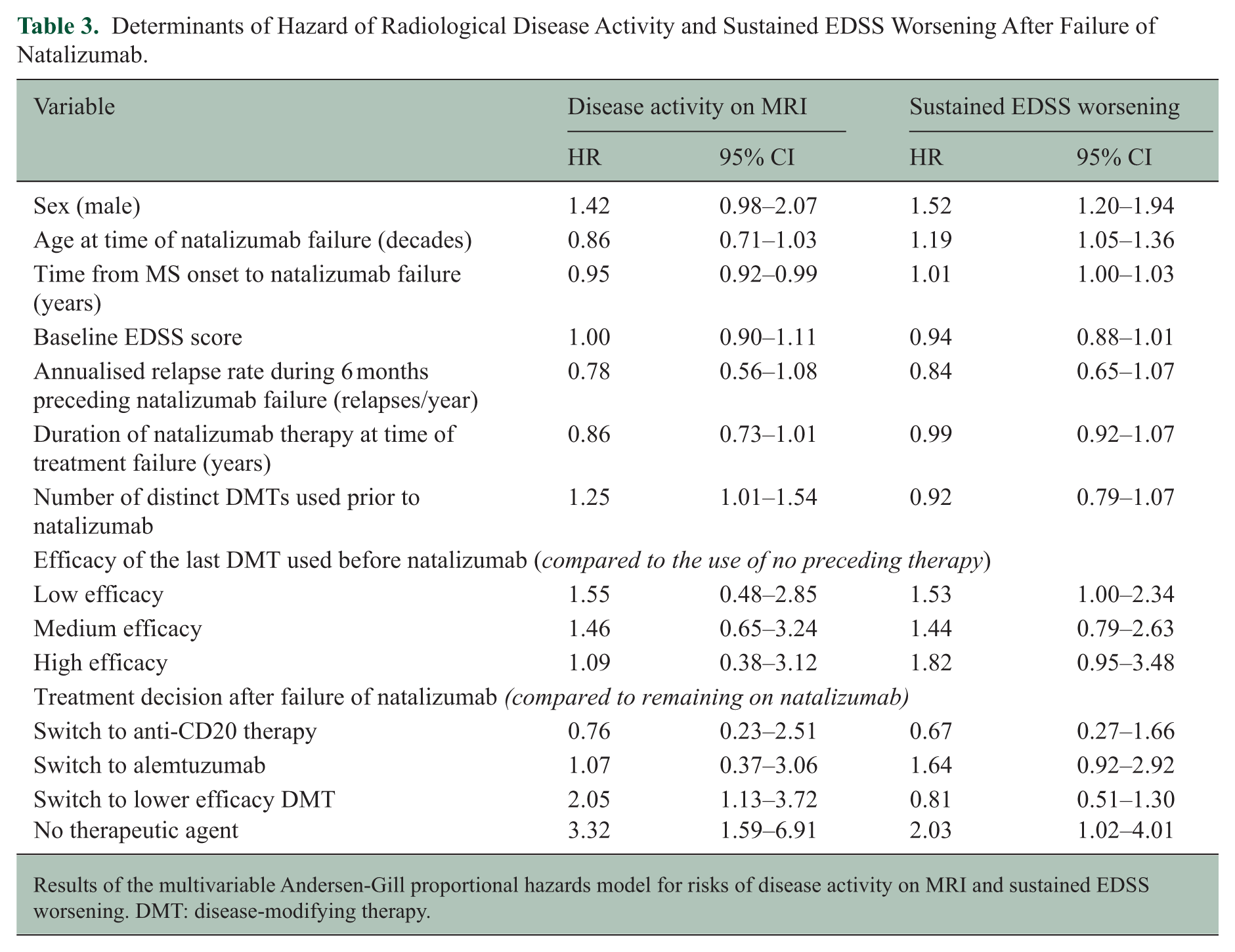
Results of the multivariable Andersen-Gill proportional hazards model for risks of disease activity on MRI and sustained EDSS worsening. DMT: disease-modifying therapy.
Sustained EDSS worsening
Results are displayed in Table 3. There were 329 events of sustained EDSS worsening during follow-up (Supplemental Table S5). Untreated periods were associated with an increased risk of EDSS worsening (HR = 2.03, 95% CI = 1.02–4.01). There was no evidence for an association between switching to anti-CD20 therapy or alemtuzumab and the risk of EDSS worsening (HR = 0.67, 95% CI = 0.27–1.66; HR = 1.64, 95% CI = 0.92–2.92, respectively). Pre-natalizumab use of low-efficacy DMTs was associated with increased risk of EDSS worsening compared to no preceding treatment (HR = 1.53, 95% CI = 1.00–2.34). Male sex and older age at time of natalizumab failure were associated with an increased risk of EDSS worsening (HR = 1.52, 95% CI = 1.20–1.94; HR = 1.12, 95% CI = 1.05–1.36 per decade, respectively).
No evidence of disease activity
Event numbers are reported in Supplemental Table S5. Results are displayed in Supplemental Table S7. Periods of no DMT therapy were associated with reduced disease-activity-free survival (HR = 2.32, 95% CI = 1.34–4.04). There was no definitive evidence to support an association between switching to other high-efficacy DMTs and a change in disease activity-free survival, though coefficients were consistent with the primary analysis (HR = 0.50, 95% CI = 0.23–1.07 for switch to anti-CD20). Increased annualised relapse rate preceding natalizumab failure was associated with reduced disease-activity-free survival (HR = 1.29, 95% CI = 1.10–1.51). Longer duration of natalizumab therapy before baseline was associated with a reduction in the risk of disease activity.
Sensitivity analyses
Results of the sensitivity analyses are displayed in Supplemental Tables S8 and S9. The association between switching to anti-CD20 therapies and reduced risk of subsequent relapses, compared to continuing natalizumab treatment, was replicated across all four analyses (HR = 0.19, 95% CI = 0.05–0.78 for patients treated with natalizumab for ⩾12 months before baseline; HR = 0.55, 95% CI = 0.31–0.97 for patients on no high-efficacy therapy before natalizumab; HR = 0.50, 95% CI = 0.29–0.86 for patients who experienced a relapse or MRI activity while treated with natalizumab; HR = 0.46, 95% CI = 0.23–0.93 when only considering relapses requiring treatment or rated as moderate/severe). Again, we found no evidence for a difference in relapse risk between switching to alemtuzumab and continuing natalizumab. The direction and the magnitude of the associations for treatment de-escalations and periods of no treatment were consistent with the primary analysis.
Discussion
In this study from the international MSBase registry, we compared outcomes among patients with different treatment paths following relapses during treatment with natalizumab. According to our results, switching to anti-CD20 therapies (ocrelizumab or rituximab) is associated with a greater than 50% reduction in the risk of subsequent relapses, in comparison with continuing natalizumab therapy. No differences were observed in exploratory secondary outcomes (MRI activity, EDSS worsening and disease-activity-free survival), noting limitations in power and trends consistent with the primary outcome. This association was not observed in those who switched therapy to alemtuzumab, noting low patient numbers. Our conclusions are supported by multiple sensitivity analyses.
Expectedly, a higher relapse rate preceding natalizumab failure predicted a higher risk of subsequent relapses. This was also the case for increased DMT use prior to natalizumab, likely reflecting more severe underlying disease. Meanwhile longer duration of natalizumab treatment before failure was associated with a lower risk of subsequent relapses, likely reflecting a better overall treatment response. Switching to anti-CD20 therapies following natalizumab was associated with a 52% reduction in risk of subsequent relapses compared to those who remained on natalizumab in the adjusted multivariable models. Based on only minor differences in the effectiveness of ocrelizumab and rituximab, which do not bear clinical significance, we have combined these therapies into the anti-CD20 group; 27 when analysed individually, hazard ratios were near-identical for ocrelizumab and rituximab. Confirming this finding was the expected increase in relapse risk following switches to lower-efficacy DMTs (46%) or during periods of no treatment (108% risk increase), in comparison with continuing natalizumab. We note that lower-efficacy DMTs were compared as a heterogeneous group, and conclusions should not be drawn regarding any individual agent in this group. Switching to alemtuzumab did not improve outcomes when compared with continuing natalizumab, though this result may be due to underpowering and should be interpreted cautiously.
Escalation to high-efficacy DMTs has consistently shown an association with improved outcomes in patients who experience disease activity despite treatment with low- and medium-efficacy DMTs,11,12,28 which is reflected by current MS treatment guidelines.7,29 However, following high-efficacy DMT failure, the only escalation treatment strategy with evidence for increased effectiveness is AHSCT, a therapy with significant toxicity.13,30 Whether switching to another class of high-efficacy DMT is a favourable treatment strategy in this situation was not clear based on current literature. Differences in the effectiveness of natalizumab and anti-CD20 therapies are small based on current evidence,3 –6 with further studies in larger cohorts required. Similarly, the effectiveness of alemtuzumab and natalizumab was shown to be similar (with the exception of a higher probability of disability improvement on natalizumab).2,31,32 Therefore, our results imply that choosing a different mechanism of action (B-cell depletion) with similar, or potentially marginally higher overall effectiveness, leads to a better response in situations where lymphocyte sequestration by natalizumab fails to control central nervous system inflammation, and provides evidence for trialling anti-CD20 therapies prior to exposing patients to the potential risks of AHSCT. This differential response possibly reflects immunobiological heterogeneity within MS,33,34 though more work is needed to explore this hypothesis.
The secondary outcome analyses were relatively less powered and thus predominantly exploratory. In contrast to our primary outcome, where 805 relapses occurred during the follow-up period, our secondary outcomes were relatively rare. There were only 329 sustained EDSS worsening events captured. Due to its more stringent criteria for sustained disability change, EDSS worsening events are less common than relapses. Likewise, 78% of patients had ⩾1 MRI recorded during follow-up, and only 172 instances of new disease activity on MRI were captured, reflecting the scarcity of MRI data in MSBase and that MRI scans are typically performed annually, limiting the opportunity to capture disease activity radiologically within a set timeframe. Indeed, the median number of MRIs per year during follow-up (in patients with ⩾1 MRI recorded) was 1.05 (quartiles: 0.66–1.53). Considering the median follow-up (2.3 years), this explains the limited opportunity to capture MRI activity in this study. Meanwhile, relapses are recorded close to the time of occurrence and at an unrestricted frequency. Despite these limitations, the exploratory secondary analyses showed trends consistent in direction and magnitude with the primary analysis.
The main limitation of this study is its observational nature. However, a randomised controlled trial specifically addressing treatment decision following natalizumab failure would be highly impractical and costly.35,36 We controlled the analyses for multiple demographic and clinical characteristics, including time-varying variables, to mitigate treatment indication bias, account for heterogeneity among participating centres, and account for treatment decisions that are time-varying. Immortal time bias was addressed by analysing treatment as a time-varying variable and setting a unified baseline definition.37,38 We only utilised prospectively acquired data to address recall bias and applied rigorous data control procedures to reduce syntactic data error. The limited number of patients who switched to another high-efficacy DMT following a relapse on natalizumab limits our available power. Clinical evidence of inflammatory activity during treatment with natalizumab is rare, 39 and most high-efficacy alternatives (ocrelizumab, alemtuzumab) have only been available in clinical use for less than a decade. Despite this limited power, our primary analysis (relapses) has found evidence of a difference between treatment groups; unfortunately, secondary outcomes were rarer and further underpowered, therefore remaining predominantly exploratory. Due to the limited opportunity for capturing MRI data, MRI outcomes only occurred in 11.7% during follow-up. Similarly, EDSS outcomes were considerably rarer than relapses. Sustained EDSS worsening occurs at an average rate of 0.41–1.14 events per 10 patient-years,17,23,40 and therefore requires longer follow-up than relapses, which occur at a rate of 0.68–1.002 relapses per year. 41 Despite this, the observed associations and trends were consistent across the analyses. The lack of data on natalizumab-neutralising antibodies presents another limitation. While neutralising antibodies could explain relapses observed on natalizumab, the access to testing for neutralising antibodies has recently become restricted, and therefore their presence or absence does not contribute to clinical decision-making in this situation. Furthermore, persisting with natalizumab treatment still led to superior outcomes compared to switching to lower-efficacy DMTs, suggesting that neutralising antibodies are unlikely to account for most cases of natalizumab failure. The number of statistical tests of hypotheses in the primary analysis was limited and did not justify correction for multiplicity. 42 Finally, there is subjectivity in the definition of a relapse in comparison with secondary outcomes. To address this limitation, a sensitivity analysis only including relapses rated as moderate/severe or requiring treatment was conducted, which replicated the primary findings; nonetheless, this remains a potential limitation of the study.
We have demonstrated that switching to anti-CD20 therapy after clinical failure of natalizumab is more than twice as effective at reducing the risk of further relapses than continuing natalizumab. No differences were demonstrated in clinically important secondary outcomes (MRI activity, disability worsening, disease-activity-free survival); however, these were exploratory in nature given limited power, with trends nonetheless favouring anti-CD20 therapies. Given these discrepancies, these results are predominantly hypothesis-generating, with larger studies needed to address these meaningful outcomes. Neurologists managing patients who experience breakthrough disease activity on natalizumab should therefore consider a switch to anti-CD20 therapy – with consideration of its individual risk-benefit profile, and noting our findings are confined to the risk of further clinical relapses. More generally, this study also shows that suboptimal response to one therapy within the high-efficacy treatment class should not preclude a switch to another high-efficacy therapy with a different mechanism of action.
Supplemental Material
sj-docx-1-msj-10.1177_13524585251398682 – Supplemental material for Managing reactivation of multiple sclerosis during treatment with natalizumab
Supplemental material, sj-docx-1-msj-10.1177_13524585251398682 for Managing reactivation of multiple sclerosis during treatment with natalizumab by Nathaniel Lizak, Sifat Sharmin, Dana Horáková, Eva Kubala Havrdova, Sara Eichau, Anneke van der Walt, Helmut Butzkueven, Jeannette Lechner-Scott, Katherine Buzzard, Olga Skibina, Oliver Gerlach, Alexandre Prat, Marc Girard, Pierre Duquette, Raed Alroughani, Francesco Patti, Francois Grand’Maison, Maria José Sá, Eduardo Aguera-Morales, Suzanne Hodgkinson, Pierre Grammond, Jens Kuhle, Bassem Yamout, Samia J Khoury, Serkan Ozakbas, Tunde Csepany, Nevin John, Guy Laureys, Murat Terzi, Maria Pia Amato, Cavit Boz, Abdullah Al-Asmi, Elisabetta Cartechini, Riadh Gouider, Saloua Mrabet, Izanne Roos and Tomas Kalincik in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors wish to thank all patients and their carers who have participated in this study and who have contributed data to the MSBase registry. The list of MSBase Study Group co-investigators and contributors is given in the online supplement. N.L., I.R. and T.K. had full access to all the data in the study, conducted the data analysis and took responsibility for the integrity of the data and the accuracy of the analysis.
Author Contributions
N.L. conducted the analysis, drafted and revised the manuscript. I.R. and T.K. edited, revised and approved the manuscript, conceptualised and designed the study, contributed substantially to data acquisition and interpreted the analysis. S.S., D.H., E.K.H., S.E., A.W., H.B., J.L.S., K.B., O.S., O.G., A.P., M.G., P.D., R.A., F.P., F.G.M., M.J.S., E.A.M., S.H., P.G., J.K., B.Y., S.J.K., S.O., T.C., N.J., G.L., L.V.H., M.T., M.P.A., C.B., A.A.A., E.C., R.G. and S.M. contributed substantially to data acquisition, interpretation of the analysis and have revised and approved the manuscript.
Data Availability
The MSBase registry is a data processor and warehouses data from individual principal investigators who agree to share their datasets on a project-by-project basis. External party access to data is subject to reasonable requests and solely at the discretion of the principal investigators. Permission for data access must be sought individually from the respective principal investigators.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article. Nathaniel Lizak received conference travel support from Merck (which owns patent rights to cladribine and Rebif (Interferon-beta-1a)). Dana Horáková was supported by the Charles University: Cooperation Programme in Neuroscience, by the project National Institute for Neurological Research (Programme EXCELES, ID Project No. LX22NPO5107) – Funded by the European Union – Next Generation EU, and by the General University Hospital in Prague project MH CZ-DRO-VFN64165. She also received compensation for travel, speaker honoraria and consultant fees from Biogen Idec, Novartis, Merck, Bayer, Sanofi Genzyme, Roche and Teva, as well as support for research activities from Biogen Idec. Eva Kubala Havrdova received honoraria/research support from Biogen, Merck Serono, Novartis, Roche and Teva; has been member of advisory boards for Actelion, Biogen, Celgene, Merck Serono, Novartis and Sanofi Genzyme; received honoraria/research support from Biogen, Merck Serono, Novartis, Roche and Teva; has been member of advisory boards for Actelion, Biogen, Celgene, Merck Serono, Novartis and Sanofi Genzyme; and has been supported by the Czech Ministry of Education – project Cooperatio LF1, research area Neuroscience and the project National Institute for Neurological Research (Programme EXCELES, ID project No LX22NPO5107) – funded by the European Union-Next Generation EU. Sara Eichau have received speaker honoraria and consultant fees from Biogen Idec, Novartis, Merck, Janssen, Bristol-Meyers, Bayer, Sanofi Genzyme, Roche and Teva. Anneke van der Walt served on advisory boards and received unrestricted research grants from Novartis, Biogen, Merck and Roche. She has received speaker’s honoraria and travel support from Novartis, Roche and Merck. She receives grant support from the National Health and Medical Research Council of Australia and MS Research Australia. Helmut Butzkueven received institutional (Monash University) funding from Biogen, F. Hoffmann-La Roche Ltd, Merck, Alexion, CSL and Novartis; has carried out contracted research for Novartis, Merck, F. Hoffmann-La Roche Ltd and Biogen; has taken part in speakers’ bureaus for Biogen, Genzyme, UCB, Novartis, F. Hoffmann-La Roche Ltd and Merck; has received personal compensation from Oxford Health Policy Forum for the Brain Health Steering Committee. Jeannette Lechner-Scott’s travel compensation from Novartis, Biogen, Roche and Merck. Her institution receives the honoraria for talks and advisory board commitment as well as research grants from Biogen, Merck, Roche, TEVA and Novartis. Katherine Buzzard received speaker honoraria and/or education support from Biogen, Teva, Novartis, Genzyme-Sanofi, Roche, Merck and Alexion; she has been a member of advisory boards for Merck and Biogen. Olga Skibina received honoraria and consulting fees from Bayer Schering, Novartis, Merck, Biogen and Genzyme. Oliver Gerlach has no disclosures to declare. Alexandre Prat has no disclosures to declare. Marc Girard has no disclosures to declare. Pierre Duquette served on editorial boards and has been supported to attend meetings by EMD, Biogen, Novartis, Genzyme and TEVA Neuroscience. He holds grants from the CIHR and the MS Society of Canada and has received funding for investigator-initiated trials from Biogen, Novartis and Genzyme. Raed Alroughani received honoraria as a speaker and for serving on scientific advisory boards from Bayer, Biogen, GSK, Merck, Novartis, Roche and Sanofi-Genzyme. Francesco Patti received personal compensation for serving on advisory board by Almirall, Alexion, Biogen, Bristol, Janssen, Merck, Novartis and Roche. He further received a research grant by Alexion, Almirall, Biogen, Bristol, Merck, Novartis and Roche and by FISM, Reload Association (Onlus), Italian Health Minister, and University of Catania. Francois Grand’Maison received honoraria or research funding from Biogen, Genzyme, Novartis, Teva Neurosciences and ATARA Pharmaceuticals. Maria José Sá received consulting fees, speaker honoraria and/or travel expenses for scientific meetings from Alexion, Bayer Healthcare, Biogen, Bristol Myers Squibb, Celgene, Janssen, Merck-Serono, Novartis, Roche, Sanofi and Teva. Eduardo Aguera-Morales has received honoraria as a speaker from Biogen, Merck, Novartis and Sanofi-Genzyme and for serving on scientific advisory boards from Novartis. Suzanne Hodgkinson has received consulting fees and speaker honoraria from Biogen, Novartis, Roche and Merck, and has received grants for her Institution from Biogen, Merck, Novartis and Roche. Pierre Grammond has served on advisory boards for Novartis, EMD Serono, Roche, Biogen idec, Sanofi Genzyme, Pendopharm and has received grant support from Genzyme and Roche. He has received research grants for his institution from Biogen idec, Sanofi Genzyme and EMD Serono. Jens Kuhle received speaker fees, research support, travel support and/or served on advisory boards by Swiss MS Society, Swiss National Research Foundation (320030_189140/1), University of Basel, Progressive MS Alliance, Alnylam, Bayer, Biogen, Bristol Myers Squibb, Celgene, Immunic, Merck, Neurogenesis, Novartis, Octave Bioscience, Quanterix, Roche, Sanofi, Stata DX. Bassem Yamout received honoraria as a speaker and member of scientific advisory boards from Sanofi, Bayer, Biogen, Merck, Janssen, Novartis, Roche and Aspen. Samia J. Khoury received compensation for serving on the IDMC for Biogen. Serkan Ozakbas has no disclosures to declare. Tunde Csepany received speaker honoraria/ conference travel support from Biogen, Merck, Novartis, Roche, Sanofi-Aventis and Teva. Nevin John NAJ is a PI on commercial MS studies sponsored by Novartis, Roche, Biogen and Sanofi. He has received speaker’s honoraria from Merck. He has had conference travel and registration reimbursement from Novartis. Guy Laureys received travel and/or consultancy compensation from Sanofi-Genzyme, Roche, Teva, Merck, Novartis, Celgene and Biogen. Murat Terzi received travel grants from Novartis, Bayer-Schering, Merck and Teva; has participated in clinical trials by Sanofi Aventis, Roche and Novartis. Maria Pia Amato received honoraria as a consultant on scientific advisory boards by Biogen, Bayer-Schering, Merck, Teva and Sanofi-Aventis; has received research grants by Biogen, Bayer-Schering, Merck, Teva and Novartis. Cavit Boz received conference travel support from Biogen, Novartis, Bayer-Schering, Merck and Teva; has participated in clinical trials by Sanofi Aventis, Roche and Novartis. Abdullah Al-Asmi received personal compensation for serving as a Scientific Advisory or speaker/moderator for Novartis, Biogen, Roche, Sanofi-Genzyme and Merck. Elisabetta Cartechini has no disclosures to declare. Riadh Gouider has received personal compensation for consulting, serving on a scientific advisory Board, speaking or other activities from Biogen, Hikma, Merck, Roche and Sanofi/Genzyme. Saloua Mrabet has received a MENACTRIMS clinical fellowship grant (2020). Izanne Roos has served on scientific advisory boards, received conference travel support and/or speaker honoraria from Roche, Novartis, Merck and Biogen. Izanne Roos is supported by MS Australia and the Trish Multiple Sclerosis Research Foundation. Tomas Kalincik served on scientific advisory boards for MS International Federation and World Health Organisation, BMS, Roche, Janssen, Sanofi Genzyme, Novartis, Merck and Biogen, steering committee for Brain Atrophy Initiative by Sanofi Genzyme, received conference travel support and/or speaker honoraria from WebMD Global, Eisai, Novartis, Biogen, Roche, Sanofi-Genzyme, Teva, BioCSL and Merck and received research or educational event support from Biogen, Novartis, Genzyme, Roche, Celgene and Merck.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This research was funded in whole or in part by the National Health and Medical Research Council (grants: 2040418, 2033165 and 2026836) and MS Australia (24-PGSR2-0129). For the purposes of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. The MSBase Foundation is a not-for-profit organisation that receives support from Roche, Merck, Biogen, Novartis, Bayer-Schering, Sanofi Genzyme and BioCSL. The study was conducted separately and apart from the guidance of the sponsors.
Ethical Considerations
The MSBase registry (registered with WHO ICTRP, ID ACTRN12605000455662) was approved by both the Melbourne Health Research Ethics Committee and local ethics committees in participating centres (as per local regulations).
Consent to Participate
Enrolled patients provided written informed consent.
Consent for Publication
Not applicable
ORCID iDs
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.