
Abstract
Background:
It is unknown whether people with aquaporin-4 antibody positive (AQP4-IgG+) neuromyelitis optica spectrum disorder (NMOSD) experience a prodrome, although a few cases report AQP4 + serology up to 16 years before the first attack.
Objectives:
To evaluate whether individuals with AQP4-IgG + NMOSD have prodromal neurologic symptoms preceding the first attack.
Methods:
We reviewed medical records of participants meeting the 2015 diagnostic criteria for AQP4-IgG + NMOSD from four demyelinating disease centres in the Canadian NMOSD cohort study CANOPTICS. We searched for neurologic symptoms occurring at least 30 days before the first attack.
Results:
Of 116 participants with NMOSD, 17 (14.7%) had prodromal neurologic symptoms. The median age was 48 years (range 25–83) at first attack; 16 (94.1%) were female. Participants presented with numbness/tingling (n = 9), neuropathic pain (n = 5), visual disturbance (n = 4), tonic spasms (n = 2), Lhermitte sign (n = 2), severe headache (n = 2), incoordination (n = 2), weakness (n = 1), psychosis (n = 1) or seizure (n = 1). Of eight who underwent magnetic resonance imaging (MRI) brain, orbits and/or spinal cord, five had T2 lesions. Within 1.5–245 months (median 14) from the onset of prodromal neurologic symptoms, participants experienced their first NMOSD attack.
Conclusions:
One in seven people with NMOSD experienced neurologic symptoms before their first attack. Further investigation of a possible NMOSD prodrome is warranted.
Keywords
Introduction
A prodrome is a set of early signs or symptoms that precedes the characteristic manifestations of disease. Parkinson disease, Alzheimer’s disease and multiple sclerosis (MS) are neurologic diseases with recognized prodromes.1–6 Common symptoms during the MS prodrome include cognitive impairment, fatigue, various types of pain, anxiety, depression, bladder dysfunction and headache.6–10 There has also been biological evidence of axonal degeneration in advance of MS clinical onset, as demonstrated by elevated levels of neurofilament light chain (NfL), in serum samples collected a median of 6 years before the first clinical attack. 11
Neuromyelitis optica spectrum disorder (NMOSD) is a relapsing inflammatory disease of the central nervous system (CNS) associated with aquaporin-4 immunoglobulin G antibodies (AQP4-IgG).12,13 Attacks frequently involve the optic nerves or spinal cord and result in significant disability with poor recovery. 14 Unlike MS, it is unknown whether people with NMOSD experience a prodromal phase. However, several cases reported in the literature indicate presence of the pathogenic AQP4-IgG antibody detected from serum samples collected years before the first NMOSD attack. For example, one woman with her first NMOSD clinical attack at age 34 years had three blood samples from 10 years before onset that were retrospectively tested – all were found to be positive for AQP4-IgG. 15 A cohort of 16 individuals with both NMOSD and myasthenia gravis included 4 individuals with AQP4-IgG detectable between 4 and 16 years before disease onset. 16 In all four cases, the clinical onset of NMOSD was preceded by the diagnosis of myasthenia gravis; however, it is unknown whether participants had other early neurologic symptoms that may have been atypical for myasthenia. Another report described a 12-year-old with asymptomatic optic disc oedema detected on routine examination. Magnetic resonance imaging (MRI) revealed evidence of a longitudinally extensive optic neuritis and serum was positive for AQP4-IgG. 17 These reports suggest that the pathogenesis of NMOSD could begin before the first typical attack. In addition, there has been growing evidence of subclinical disease activity in established NMOSD, including asymptomatic MRI lesions, microstructural visual system changes and elevated glial fibrillary acidic protein (GFAP) levels in some individuals between attacks.18–21
Comprehensive investigations into prodromal symptoms in the years before the first NMOSD attack are lacking. There have been several case series and case reports of symptoms in the days or weeks immediately before disease onset with some reports limited to infectious triggers of a first attack.22–25 Recognition and characterization of NMOSD prodromal symptoms could have implications for our understanding of the pathogenesis of the disease, as well as its diagnosis and management. Importantly, detection of early features may support prompt diagnosis and greater ability to prevent neurologic disability.
Our aim was to evaluate the presence of prodromal neurologic symptoms in people with AQP4-IgG-positive NMOSD before the first NMOSD attack. We hypothesized that people with AQP4-IgG-positive NMOSD would have varying prodromal neurologic symptoms. Based on clinical experience, we speculated that sensory symptoms and pain may be part of an NMOSD prodrome. We reviewed the medical records of NMOSD participants enrolled in the multicentre observational Canadian NMOSD and other atypical demyelinating diseases cohort study (CANOPTICS).
Materials and methods
Study design, setting and participants
Eligible participants were selected from those enrolled in CANOPTICS at four Canadian demyelinating disease centres: St. Michael’s Hospital (Toronto), Sunnybrook Health Sciences Centre (Toronto), the Ottawa Hospital (Ottawa) and the University of Manitoba (Winnipeg). For inclusion, participants were required to have a diagnosis of AQP4-IgG-positive NMOSD by 2015 International Panel on NMO Diagnosis (IPND) criteria. 12 AQP4-IgG status was determined by serum samples using a cell-based or enzyme linked immunosorbent assay (ELISA) assay. As per the CANOPTICS protocol, participants were 18 years or older at the time of recruitment, but the first attack was evaluated retrospectively and could occur at any age. Data of interest were retrospectively collected from the participants’ electronic medical records and CANOPTICS REDCap database. This study was approved by the Medical Ethics Committee of each centre and conducted in accordance with recognized ethical standards. Written informed consent was obtained from all participants.
Evaluation of clinical and imaging features
Prodromal neurologic symptoms were considered where symptoms occurred at least 30 days before the first typical NMOSD attack diagnosed by the treating neurologist and meeting 2015 IPND criteria. The 30-day cutoff was chosen to exclude symptoms that could have been directly related to the first NMOSD attack. We omitted episodes where the diagnosis of NMOSD was delayed, but in retrospect the symptoms and imaging features were consistent with an attack meeting 2015 IPND criteria. The presence of systemic prodromal symptoms was also documented where reported.
Prodromal neurologic symptoms were reviewed for clinical features (symptom quality and severity), timing (date of onset relative to the first attack, frequency, duration), outcome and associated radiological findings. It was noted whether participants had one or more than one type of symptom. Ophthalmology records were reviewed when available for participants with visual symptoms. We documented the time from the onset of the first prodromal neurologic symptom to the first NMOSD attack. Radiological reports were collected including computerized tomography (CT) head and MRI of the brain, orbits and/or spinal cord obtained at the time of the prodromal neurologic symptoms and at the time of first NMOSD attack. All cases were screened by at least one neurologist with expertise in demyelinating disease. Cases with prodromal neurologic symptoms, including their radiological findings, were reviewed by one neurologist with expertise in demyelinating disease and one senior neurology resident. A structured data collection form was used.
Baseline characteristics were noted from the time of first NMOSD attack onset including age, sex, ethnicity (Asian, African or Caribbean, European or White, Latin, Indigenous or unknown), pre-existing autoimmune disease (excluding thyroid disease), pre-existing malignancy and clinical phenotype of the first NMOSD attack (transverse myelitis, optic neuritis, area postrema syndrome, cerebral syndrome, brainstem or cerebellar syndrome, diencephalic syndrome and multifocal).
Statistical analysis
We analysed baseline characteristics at the time of the first NMOSD attack for three groups: (1) the whole cohort, (2) participants with prodromal neurologic symptoms and (3) participants without prodromal neurologic symptoms. For those with prodromal neurologic symptoms, we described quality, timing, duration and the outcome of such symptoms, and imaging features where available. Descriptive statistics were reported as frequency counts and percentages for categorical variables, and as median and range for continuous variables. We compared the differences in baseline characteristics between those with and without prodromal symptoms using chi-square test and Fisher’s exact test for categorical variables and Wilcoxon rank-sum test for continuous variables. We set p < 0.05 as being statistically significant. We used Stata Statistical Software: Release 14 (StataCorp LP, College Station, TX, USA).
Results
Of 116 participants with AQP4-IgG-positive NMOSD, 17 (14.7%) had prodromal neurologic symptoms. The median age at first NMOSD attack was 48 years (range 25–83) and 16 were female (94.1% ratio 16:1). Participants experienced a variety of first NMOSD attacks, including transverse myelitis (n = 10), optic neuritis (n = 4), area postrema syndrome (n = 1), cerebral syndrome (n = 1) and brainstem or cerebellar syndrome (n = 1); participants with prodromal symptoms compared with those without were more likely to present with transverse myelitis as a first attack and less likely to present with optic neuritis. Other baseline characteristics for participants with prodromal neurologic symptoms were generally similar to those without prodromal neurologic symptoms, although there was a relative preponderance of women and people of African or Caribbean ethnicity (Table 1).
Baseline characteristics at first NMOSD attack.
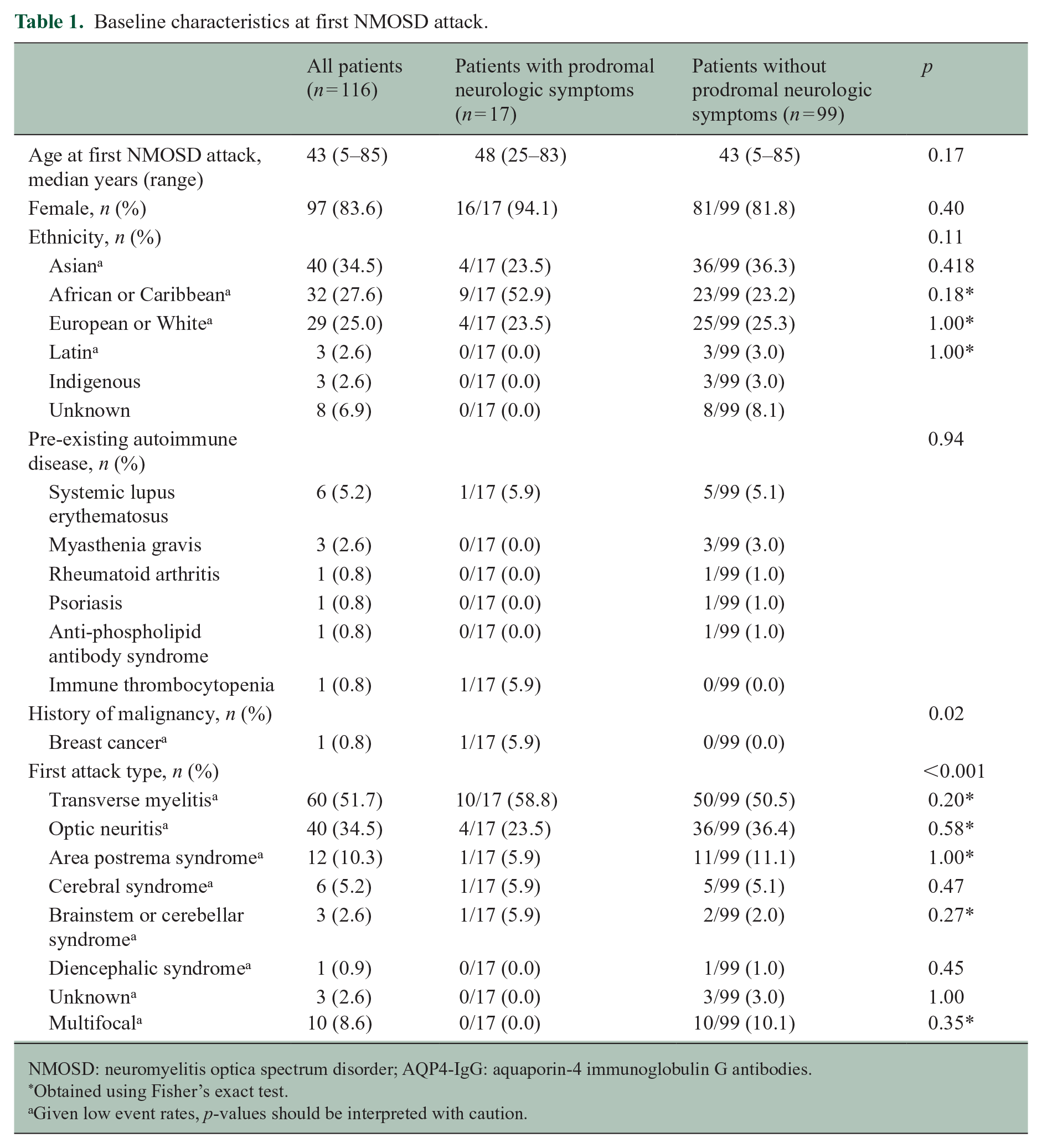
NMOSD: neuromyelitis optica spectrum disorder; AQP4-IgG: aquaporin-4 immunoglobulin G antibodies.
Obtained using Fisher’s exact test.
Given low event rates, p-values should be interpreted with caution.
Participants developed prodromal neurologic symptoms a median of 14 months (range 1.5–245 months) before their first NMOSD attack. Of those with prodromal neurologic symptoms, half had numbness and/or tingling, nearly one-third had neuropathic pain and nearly one-quarter had visual disturbance. Other symptoms were rare (Table 2). Ten (58.8%) experienced more than one neurologic symptom. Symptoms were either constant (n = 10) or intermittent (n = 9). Six (35.3%) participants had symptoms that were still ongoing at the time of the first NMOSD attack (cases 1, 4, 8, 9, 11 and 17). Three (17.6%) had no recovery and three (17.6%) had partial recovery of symptoms. Twelve (70.6%) had complete resolution of symptoms before the first NMOSD attack, with a median symptom duration of 4 weeks (range 2 minutes–16 weeks). Four (23.5%) had a prodromal event that was highly consistent with their first NMOSD attack (Cases 8, 10, 13 and 15), such as prodromal tonic spasms followed by a first attack of longitudinally extensive transverse myelitis. In these cases, the median time between onset of prodromal neurologic symptoms and the first NMOSD attack was 18.5 months (range 12–92 months). Of those with prodromal neurologic symptoms, three (17.6%) participants had one or more prodromal systemic symptom including decreased appetite (n = 1), severe fatigue (n = 1), epigastric pain (n = 1) and exacerbation of underlying obstructive lung disease (n = 1). Individual cases are outlined in Table 3.
Summary of prodromal symptoms.
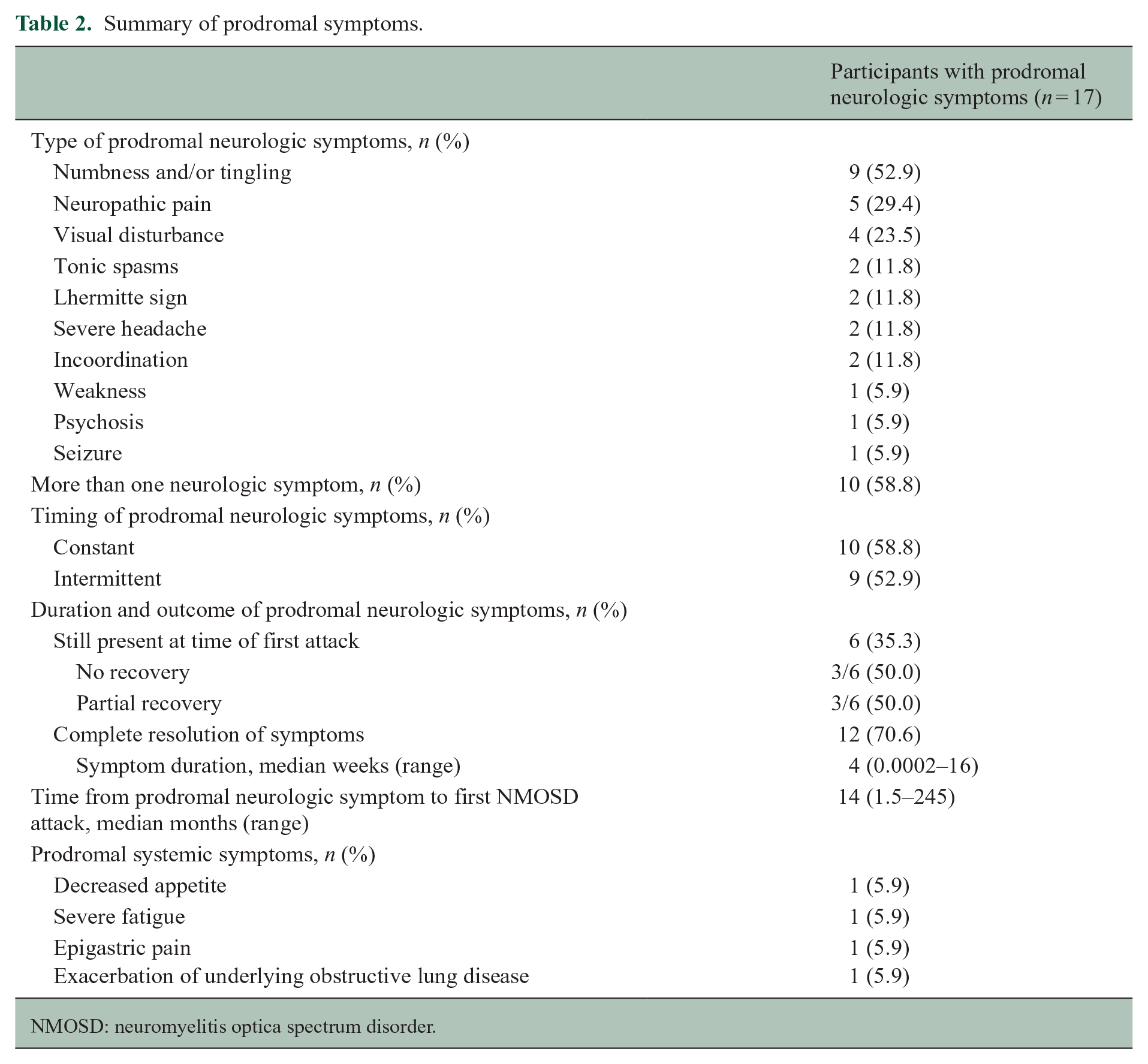
NMOSD: neuromyelitis optica spectrum disorder.
Demographic and clinical data for patients with prodromal neurologic symptoms.
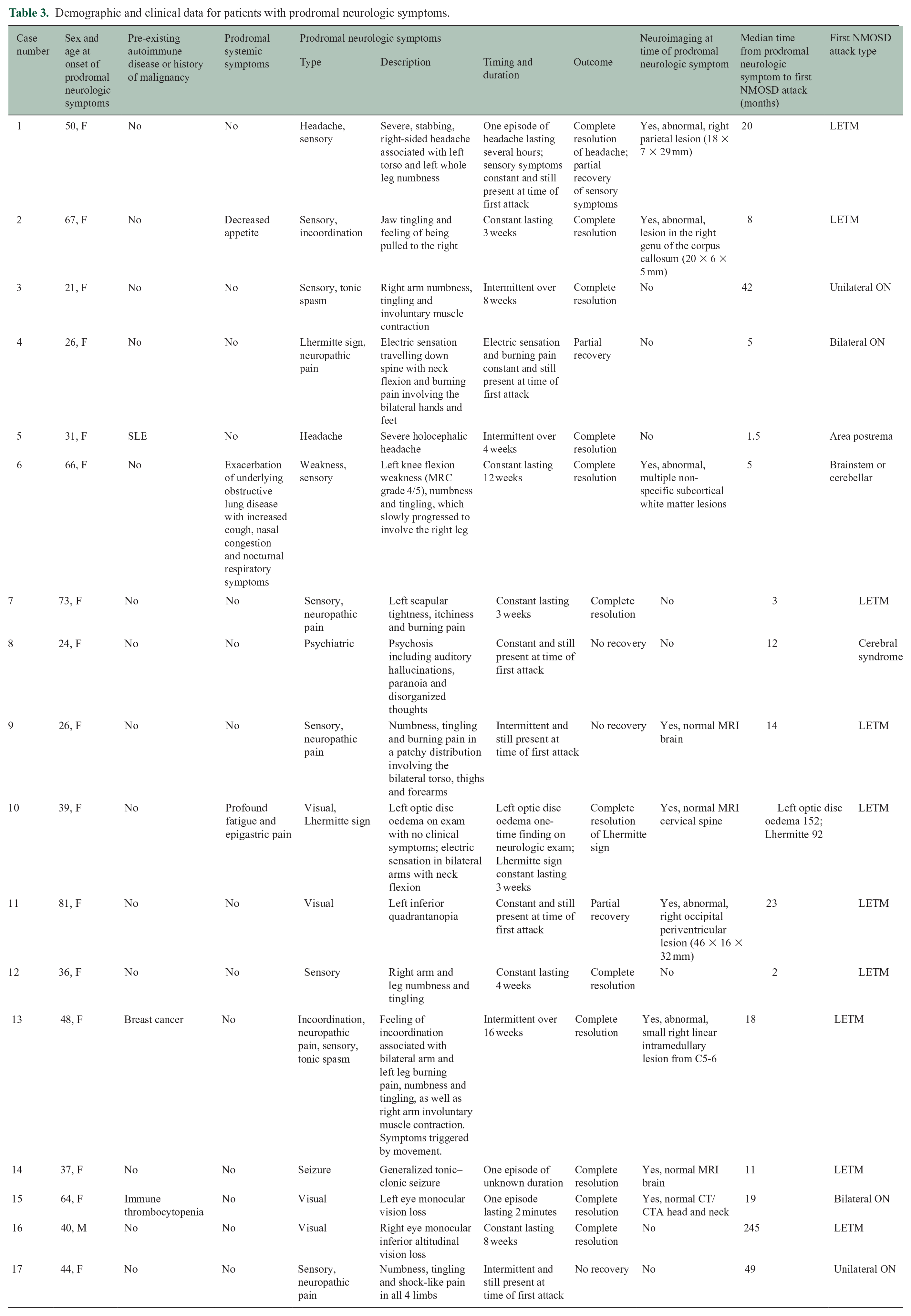
Twelve (70.6%) participants underwent one or more neuroimaging studies at the time of their prodromal neurologic symptoms (Table 4). Of seven participants who underwent CT head, one had an abnormal finding with a hypodense lesion in the right occipital periventricular region. Four out of seven participants (57.1%) had abnormal findings on MRI brain. These included the participant with the abnormal CT head, with the same right occipital periventricular lesion appearing hyperintense on T2 sequences with subtle patchy enhancement and mild diffusion restriction (Case 11; Figure 1; 3.a–d). Of the remaining three participants with abnormal MRI brain, one had a T2 hyperintense right parietal lesion (Case 1; Figure 1; 1.a–b), one had a T2 hyperintense lesion in the right genu of the corpus collosum with enhancement (Case 2) and one had non-specific T2 hyperintense subcortical white matter lesions (Case 6). One out of four participants had an abnormal MRI spine, revealing a small, linear, right intramedullary T2 hyperintense lesion from C5 to C6 with no enhancement (Case 13). Of the five participants with abnormal imaging findings, the findings in three were felt to potentially explain their prodromal neurologic symptoms (Cases1, 11 and 13). In each of these cases, the lesion did not have a typical appearance for NMOSD. 12
Neuroimaging at time of prodromal neurologic symptoms.
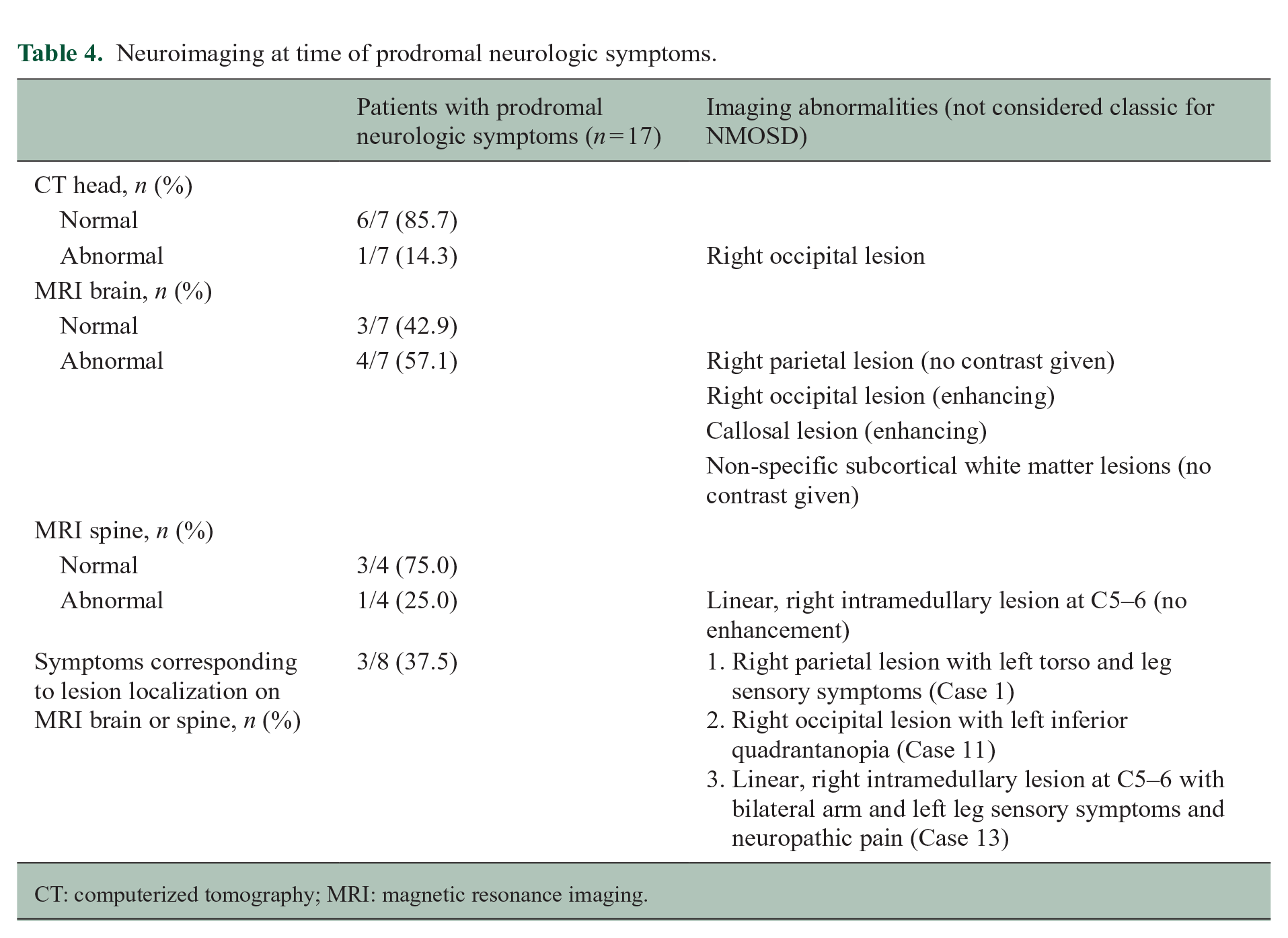
CT: computerized tomography; MRI: magnetic resonance imaging.
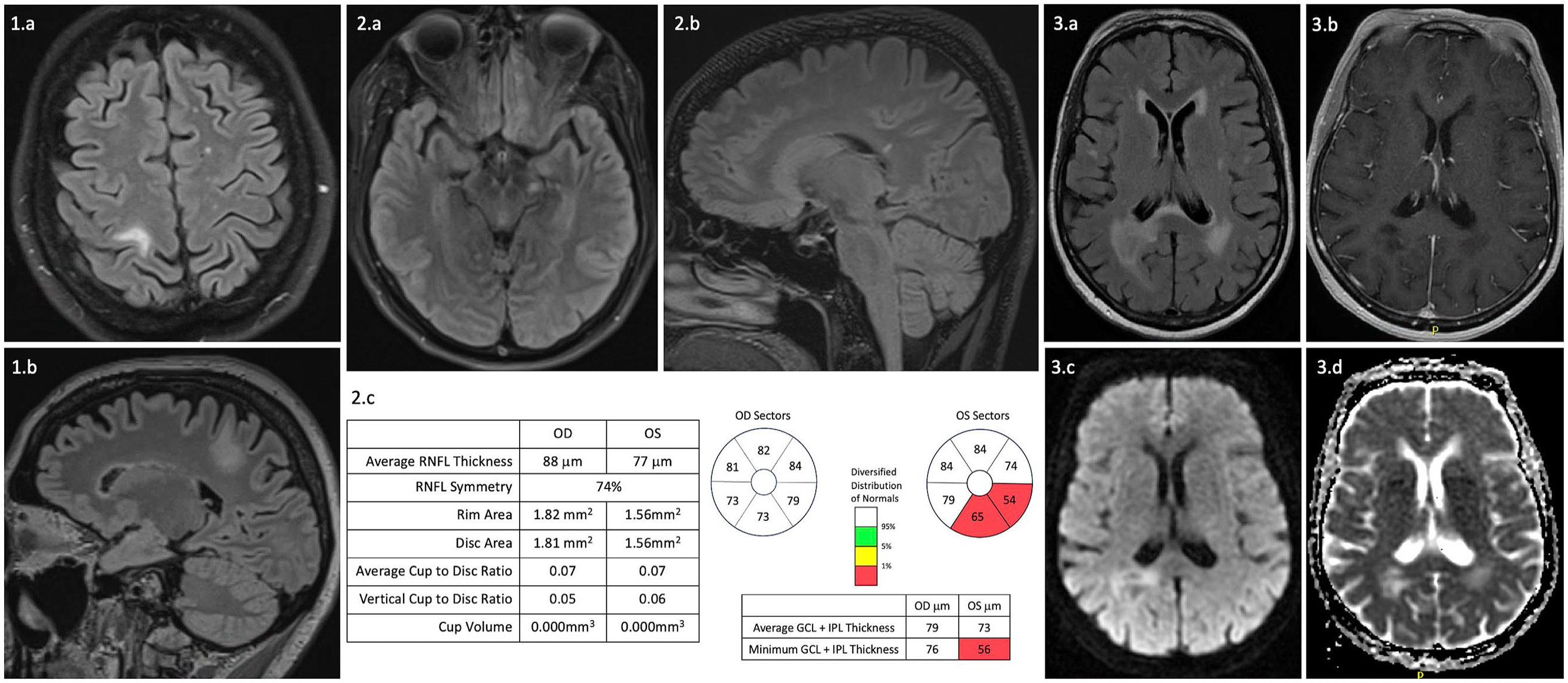
Select MRI brain and spinal cord images and optical coherence tomography in patients with prodromal neurologic symptoms preceding a diagnosis of AQP4-IgG-positive NMOSD. 1: Case 1, patient with severe right-sided headache and left torso and leg sensory symptoms. (1.a) Axial and (1.b) Sagittal T2 FLAIR show a T2 hyperintense lesion in the right parietal white matter, sparing the cortex. 2: Case 10, patient with prodromal Lhermitte sign and asymptomatic optic disc oedema. (2.a) Axial and (2.b) Sagittal T2 FLAIR at the time of the first NMOSD attack reveal T2 hyperintense lesions in the left cerebral peduncle and splenium of the corpus collosum. (2.c) Optical coherence tomography after asymptomatic left optic disc oedema shows retinal ganglion cell layer thinning in the left eye. 3: Case 11, patient with left inferior quadrantopia. (3.a) Axial T2 FLAIR and (3.b) Axial T1 post-contrast at the time of prodromal neurologic symptoms show a right occipital periventricular lesion with subtle patchy enhancement. (2.c) Diffusion weighted imaging and (2.d) Apparent diffusion coefficient show partial diffusion restriction.
All participants with prodromal neurologic symptoms had MRI brain or spinal cord at the time of the first NMOSD attack (Table 5). Eleven (64.7%) had asymptomatic lesions in the brain (n = 9), optic nerve (n = 1) or spinal cord (n = 1). The asymptomatic lesions at the time of the first attack were thought to explain the prodromal neurologic symptoms in an additional two participants (18.2%). One participant had prodromal symptoms involving neuropathic pain and Lhermitte sign, with no imaging at the time of these symptoms. She then presented with a first attack of bilateral optic neuritis 5 months later. MRI spine at the time of her presentation with bilateral optic neuritis showed a T2 hyperintense lesion from T1 to T3, as well as an isolated lesion at T6 (Case 4). The other participant had asymptomatic left optic disc oedema 13 years before presenting with a first attack of transverse myelitis. MRI brain at the time of first attack revealed subtle left optic nerve atrophy (Case 10; Figure 1; 2.a).
Neuroimaging at time of first NMOSD attack.

MRI: magnetic resonance imaging.
Of the four participants with visual disturbance, two underwent ophthalmology assessment at the time of their symptoms. The first participant had left inferior quadrantanopia without any evidence to suggest optic neuritis on ophthalmologic examination (Case 11). The second participant had left optic disc oedema on examination by her neurologist. She later underwent optical coherence tomography (OCT) which revealed left inferior ganglion cell layer thinning (Case 10; Figure 1; 2.c). This participant also experienced Lhermitte sign as a prodromal symptom and this was the primary reason for her inclusion in the study.
Discussion
A total of 1 in 7 participants out of 116 with AQP4-IgG-positive NMOSD experienced prodromal neurologic symptoms preceding the first NMOSD attack in this multicenter Canadian cohort. To our knowledge, this represents the first cohort study investigating prodromal symptoms in the years predating NMOSD. Our results, including abnormal MRI findings in certain cases, suggest that central nervous system (CNS) disease activity occurs in some individuals with NMOSD before the first typical attack. Although baseline characteristics of those who experienced prodromal neurologic symptoms were generally similar to those who did not, a relative predominance of women and participants of African or Caribbean ethnicity was observed. Notably prodromal symptoms did not appear to be accounted for by comorbidities in this cohort, as the proportions with history of autoimmune disease or malignancy were similar across groups. Further study is required to explore biomarkers such as MRI changes and serum GFAP and NfL levels that may be associated with prodromal symptoms in NMOSD. Association of prodromal neurologic symptoms with specific biomarkers may allow prodromal NMOSD to be identified, monitored, and potentially treated in the future to prevent a first disabling attack.
The observation that two-thirds of those with prodromal neurologic symptoms in this cohort underwent neuroimaging suggests that the treating physician felt the symptoms were enough to warrant investigation. Five of eight (62.5%) investigated with MRI brain, orbits and/or spine had abnormalities which were not classic for an NMOSD attack, including subcortical white matter lesions, a small lesion in the genu of the corpus callosum and a linear, short-segment spinal cord lesion. Enhancement of some of these lesions suggests that they appeared around the same time as the prodromal symptoms. In three cases, the prodromal neurologic symptoms localized to the lesion(s) identified on imaging. Thus, in some NMOSD patients, prodromal neurologic symptoms may be a reflection of mild CNS disease activity. Moreover, we observed asymptomatic optic disc oedema in one case, which was later associated with supportive findings on OCT and evidence of left optic nerve atrophy on MRI brain. In addition, there may be cases of prodromal neurologic symptoms in NMOSD that are not associated with MRI changes but could still reflect CNS inflammation.
There is evidence from studies of patients with established NMOSD that asymptomatic MRI lesions may occur during or between attacks, although the frequency has varied widely across studies, which may be in part due to whether patients were evaluated during a clinically quiescent phase and whether they were on therapy.26–28 Similarly, elevations in serum GFAP levels have been observed in some patients between attacks and may portend an increased risk of future attacks. 29 We did not have access to any serum samples collected at the time of the prodromal neurologic symptoms to evaluate GFAP and NfL levels, biomarkers associated with astroglial and neuronal pathology, but these tests would be useful to conduct in future cases, as well as testing for presence of the AQP4-IgG antibody.
The most common prodromal neurologic symptoms in this cohort were sensory symptoms and pain. These symptoms are non-specific but their character may be of relevance in flagging concern for prodromal NMOSD as opposed to another cause. Pain was typically neuropathic, described as severe, burning, electric and itching. Severe neuropathic pain is a hallmark of established NMOSD with estimated prevalence of >70% and increased frequency and severity compared with MS. 30 Tonic spasms, which were reported in two of our cases, are considered characteristic of NMOSD and in one study, were associated with an odds ratio of 22 comparing NMOSD to MS. 31 A Lhermitte sign should raise the question of a cervical cord demyelinating process even in the absence of other symptoms. Seizures have been reported as the first clinical sign of MS and MOGAD, but AQP4 + NMOSD should also be considered in the differential diagnosis. 32 Systemic prodromal symptoms reported in this study included fatigue, decreased appetite, epigastric pain, and exacerbation of obstructive lung disease, although we suspect that such symptoms may have been underreported by both participants and the treating neurologists, and warrant further investigation. There have been reports of elevated creatine kinase (CK) levels and myopathy preceding onset of NMOSD and in a large Chinese NMOSD registry there were four cases of pregnancy loss in the months preceding NMOSD onset.33,34 AQP4 is known to occur in high concentrations in the skeletal muscles, placenta and kidneys in addition to the brain, which might explain prodromal symptoms localizing to these tissues.
Strengths of this study include a detailed review of medical records and neuroimaging to identify neurologic symptoms preceding the first NMOSD attack from a multicentre, ethnically diverse cohort. Limitations include the retrospective nature of data collection, and thus the variability of the assessments that occurred including clinical documentation and neuroimaging. Participants may not have been asked about prior neurological symptoms as part of their assessment, resulting in a likely under-ascertainment of prodromal symptoms. There is also potential selection bias, as only individuals enrolled in CANOPTICS were eligible to participate in the study. Documentation of non-neurologic symptoms was sparse, and neuroimaging was only performed in a share of cases. In addition, formal ophthalmologic data were limited for participants with prodromal visual disturbance. The exclusion of symptoms that occurred within 30 days prior to the first NMOSD attack may also underestimate the number of individuals with prodromal neurologic symptoms. For a few of our cases, the prodromal neurologic symptoms could be interpreted as the early stages of the first attack or a missed first attack, as has been suggested with the MS prodrome. 35 However, if they were attacks, they did not demonstrate one of the core clinical characteristics of NMOSD by 2015 IPND criteria and raise the question of whether the clinical spectrum of NMOSD warrants further expansion. 12 It is possible that a first attack could evolve over more than 30 days, especially in participants who experienced a prodrome that was consistent with their first NMOSD attack. However, we found only two participants who had symptoms 60 days or less before the first attack. It is difficult to determine whether reported prodromal symptoms are related to the NMOSD disease process or if they represent a separate entity, but further biomarker studies may provide answers.
In conclusion, we found that 15% of this cohort with AQP4-IgG-positive NMOSD presented with prodromal neurologic symptoms before the first NMOSD attack. Participants experienced a variety of symptoms with diverse imaging findings. Prodromal neurologic symptoms did not appear to be related to comorbidities and while symptoms were non-specific, their character was typical for NMOSD. Our findings suggest that participants with NMOSD may not always manifest with neurologic symptoms, fulfilling the core clinical characteristics and associated imaging features from 2015 IPND diagnostic criteria at initial presentation. Further systematic investigation is needed to better understand the relationship between prodromal symptoms, imaging and fluid biomarkers, and the development of NMOSD.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S.L. has nothing to declare. R.A.M. receives research funding from CIHR, MS Canada, Crohn’s and Colitis Canada, National Multiple Sclerosis Society, CMSC, the Arthritis Society and the US Department of Defense, and is a co-investigator on studies receiving funding from Biogen Idec and Roche Canada. G.F. has served as a member of the advisory board for Horizon Therapeutics. M.S.F. has received research or educational grants from Sanofi-Genzyme Canada. He has received consulting fees or honoraria from Alexion/Astra Zeneca, Biogen Idec, EMD Inc./EMD Serono/Merck Serono, Find Therapeutics, Hoffman La-Roche, Horizon Therapeutics, Novartis, Quanterix, Sanofi-Genzyme, Teva Canada Innovation. He has served as a member of a company advisory board, board of directors or other similar group for Alexion/Astra Zeneca, Actelion/Janssen (J&J), Atara Biotherapeutics, Bayer Healthcare, Celestra Health, EMD Inc./Merck Serono, Find Therapeutics, Hoffman La-Roche, Novartis, Sanofi-Genzyme, Setpoint Medical. He has been part of a speaker’s bureau for Hoffman La-Roche, Novartis, and EMD Inc. L.L. is a site investigator for studies funded by Roche, Novartis and Sanofi Aventis. He has received consultation fees from Alexion, Biogen, Bristol Myers Squibb, EMD Serono, Novartis, Roche and Sanofi Aventis. A.M. has received compensation for serving on an advisory board or data safety monitoring board for Biogen, Novartis, EMD Serono and Alexion. M.V.V. has received salary support as a co-investigator from National MS Society. A.K. has nothing to declare. D.L.R. has received research funding from MS Canada, the National MS Society, CMSC, University of Toronto Division of Neurology and Roche Canada. She has received speaker or consultant fees from Alexion, Biogen, EMD Serono, Horizon Therapeutics, Novartis, Roche, Sanofi Aventis and Touch IME.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study received funding from MS Canada (EGID 918124).