
Abstract
A 52-year-old man experienced two seizures in January and June 2021. In October, the neurological examination did not reveal sensory/motor deficits. Brain magnetic resonance imaging (MRI) showed hyperintense lesions with contrast enhancement (CE) involving white matter bilaterally, brainstem, and cerebellum. Spine MRI showed hyperintense C2-C3 and C4-C6 lesions with CE. Anti-aquaporin-4 (AQP4) antibodies were detected, confirming the diagnosis of neuromyelitis optica spectrum disorder (NMOSD). The patient experienced a status epilepticus compatible with Epilepsia Partialis Continua treated with antiseizure medications. He was also treated with methylprednisolone, plasma exchange, and rituximab. Status epilepticus can be a rare manifestation of NMOSD, heightening the broad spectrum of AQP4 autoimmunity.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD) is an antibody-mediated disease primarily affecting the astrocytes and is characterized by the predominant involvement of the optic nerve and spinal cord. Transverse myelitis episodes in patients with NMOSD are characteristically longitudinally extensive (extending ⩾3 vertebral segments), and the resulting clinical phenotype is typically severe, while asymptomatic cord lesions are extremely rare.1–4 In about 80% of patients with NMOSD, immunoglobulin G autoantibodies specific to aquaporin-4 (AQP4) are detected. 1 Seizures are exceptionally rare manifestations of NMOSD occurring in <1% of the patients, and to date, there are no cases of status epilepticus (SE) in patients with NMOSD reported in the literature. 5
We herein describe a patient who developed a focal SE in association with AQP4 autoimmunity.
Case presentation
A 52-year-old man with a past medical history of hypertension and prior coronary artery bypass grafting and no family history of neurological or autoimmune diseases experienced his first seizure in January 2021. Following this episode, an electroencephalogram (EEG) and a brain computed tomography (CT) scan performed in another hospital revealed no abnormalities. Treatment with levetiracetam 1000 mg/day was started. In June 2021, the patient presented a second seizure characterized by clonic movements on the left side of the face. In October 2021, he was admitted to the neurology ward of our institution, where a neurological examination revealed slowed ideomotor function, brisk, and symmetrical reflexes in his upper limbs, Meyerson’s sign, and bilateral grasping reflex with normal motor and sensory assessment. A brain magnetic resonance imaging (MRI) showed hyperintense lesions on fluid-attenuated inversion recovery (FLAIR) and T2-weighted images, with some of them showing restriction in diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) images and contrast enhancement (CE) in T1-weighted image, involving the subcortical and periventricular white matter in both hemispheres and infratentorial lesions in the right cerebellar hemisphere and peduncle and medulla oblongata (Figure 1(a) and (b)). Spine MRI showed T2 hyperintense C2-C3 and C4-C6 lesions, both with CE (Figure 1(b) and (c)). Visual evoked potentials were unremarkable. Cerebrospinal fluid (CSF) analysis revealed increased protein content (55 mg/dL), slight pleocytosis (20 cells/mm3), and the presence of oligoclonal bands type II. An extensive panel of onconeural antibodies and antibodies against cell surface antigens, including antibodies specific to myelin oligodendrocyte glycoprotein (MOG), NMDA Receptor, GABAB Receptor, DPPX, LGI1, CASPR2, AMPAR and GABAA Receptor tested negative, AQP4 antibodies were detected in the patient’s serum by a commercial cell-based assay (EUROIMMUN, Lübeck, Germany) and confirmed by an in-house live cell-based assay as previously described, 6 supporting the diagnosis of NMOSD. 7
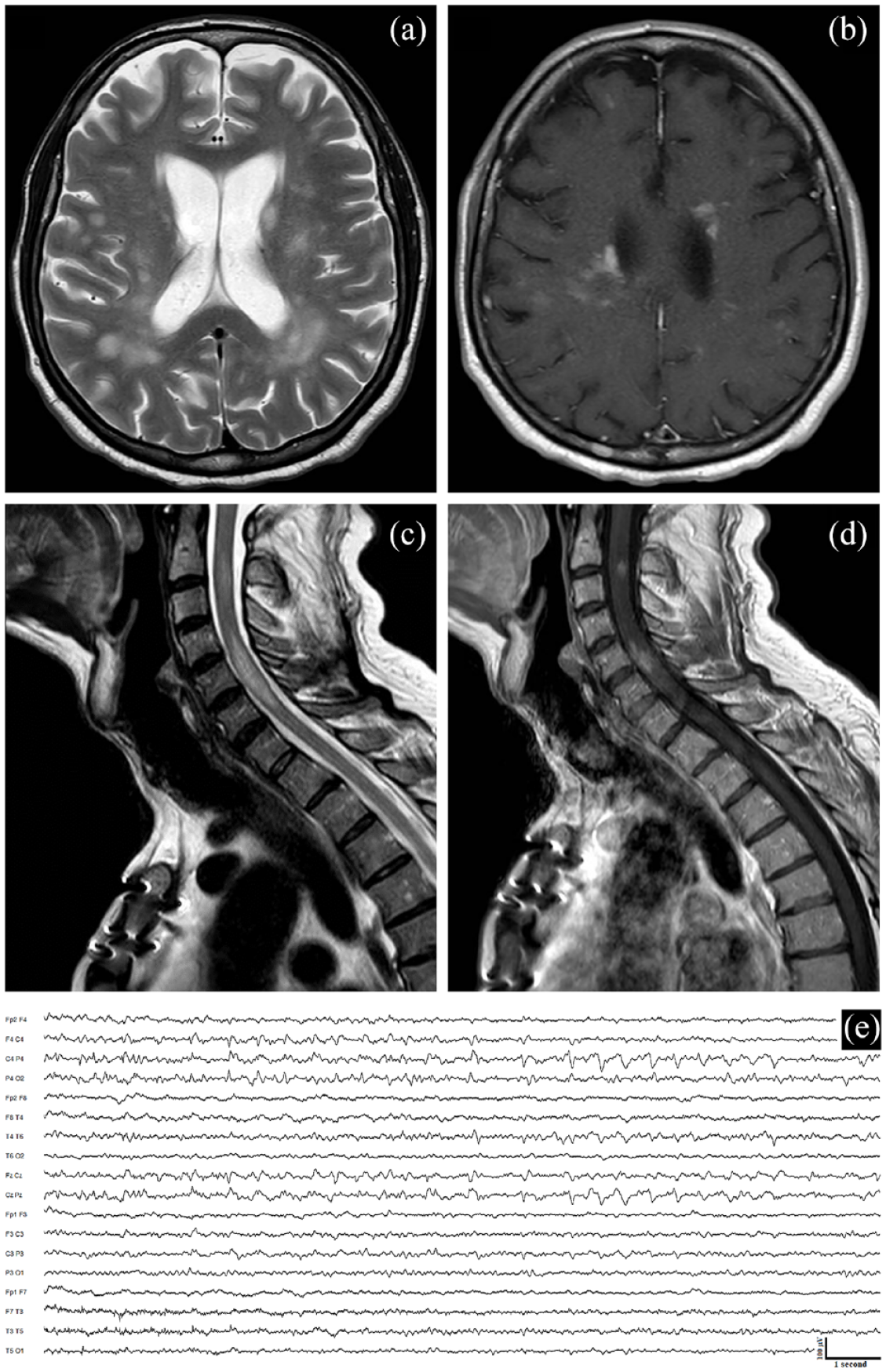
Magnetic resonance imaging (MRI) of the brain and spinal cord and ictal electroencephalogram (EEG). Brain MRI shows hyperintense lesions on T2-weighted images, with some showing cloud-like contrast enhancement after gadolinium administration involving the subcortical and periventricular white matter in both hemispheres (a and b). The spinal cord MRI reveals a C2-C3 lesion and myelitis from C4 to C6 on T2-weighted images with associated enhancement on T1 post-gadolinium images (c and d). The EEG performed during the focal motor status epilepticus (Epilepsia Partialis Continua) shows right parietal continuous high-amplitude theta/delta waves (e).
During the hospitalization, the patient experienced a focal motor SE characterized by continuous clonic movements of the left body compatible with Epilepsia Partialis Continua (EPC), the EEG during the SE revealed right parietal continuous high-amplitude theta/delta waves (Figure 1(e)).
The SE was treated with 10 mg intravenous diazepam, followed by 1600 mg of valproic acid and 750 mg of phenytoin with resolution of the SE after approximately 6 hours. The patient was then treated with intravenous methylprednisolone (1000 mg/day for 5 days, followed by a tapering scheme with prednisone 50 mg/day) combined with plasma exchange (three cycles, every other day) and was discharged on valproic acid 1000 mg/day, phenytoin 300 mg/day, and underwent rituximab infusion 1 g for two times biweekly.
At the last follow-up, 24 months later, the patient did not experience further relapses of NMOSD. Patient monitoring included peripheral memory B cells, with rituximab reinfusion scheduled upon their increase beyond 0.05% for the first 2 years and 0.1% thereafter, in line with the recommendations of Kim et al. 8 Only one focal motor seizure occurred during an attempt to taper phenytoin at the 12-month follow-up, with a brain MRI showing a reduction of the inflammatory lesions with no CE and no restricted diffusion on DWI and ADC sequences.
Discussion
The case herein described has several points of interest. This report describes an atypical case of NMOSD with a subacute onset where seizures and SE were the predominant manifestations observed during the course of the disease.
Seizures are a rare manifestation of AQP4 autoimmunity, reported in less than 1% of the cases. 1 While to the best of our knowledge, there have not been prior reports of status epilepticus being linked to NMOSD. Seizures in NMOSD are reported to occur weeks to years after the disease onset, with the seizure type described as focal, focal to bilateral tonic-clonic, or generalized tonic-clonic. 1 However, no further information regarding seizure recurrence and management is reported in the literature for these patients. Seizures occurring in the setting of the active phase of immune-mediated encephalitis are defined as acute symptomatic seizures secondary to autoimmune encephalitis, particularly in patients with autoantibodies against surface antigens that usually achieve seizure freedom after immunotherapy and in whom antiseizure medications (ASMs) can eventually be discontinued. 9 Instead, the term autoimmune-associated epilepsy suggests an enduring predisposition to seizures. 9 Acute symptomatic seizures in this context could be associated with neuronal hyperexcitability secondary to inflammation and glutamate excitotoxicity, supported by the fact that AQP4 antibodies binding to astrocytes triggers glutamate excitotoxicity secondary to antibody-mediated excitatory amino acid transporter 2 (EAAT2) internalization.10,11 On the contrary, autoimmune-associated epilepsy could be linked to gliosis, neuronal dysfunction, and synaptic abnormalities. 10
In the case of our patient, the SE can be defined as an acute symptomatic event. However, when considering the patient’s previous history of seizures and their recurrence following an attempt to reduce ASMs, our diagnosis shifts more toward the possibility of autoimmune-related epilepsy.
In addition, asymptomatic myelitis is a very unusual feature in patients with NMOSD, reported in less than 5% of the cases. In the literature, only a few cases of asymptomatic cord lesions associated with NMOSD have been documented.2,3 The cases reported had mostly short-segment lesions, and the myelitis was preceded by other manifestations, such as optic neuritis. 4
In conclusion, the present case heightens the broad spectrum of NMOSD manifestations and highlights the importance of suspicion of this disease in cases of seizures, even isolated but accompanied by compatible and suggestive MRI findings. The mainstay of treatment for seizures/epilepsy secondary to autoimmune encephalitis consists of immunotherapy along with ASMs. Further studies characterizing seizures/epilepsy in patients with AQP4 autoimmunity are required.
Footnotes
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from the patient.