
Abstract
Background:
The prognostic significance of non-disabling relapses in people with relapsing-remitting multiple sclerosis (RRMS) is unclear.
Objective:
To determine whether early non-disabling relapses predict disability accumulation in RRMS.
Methods:
We redefined mild relapses in MSBase as ‘non-disabling’, and moderate or severe relapses as ‘disabling’. We used mixed-effects Cox models to compare 90-day confirmed disability accumulation events in people with exclusively non-disabling relapses within 2 years of RRMS diagnosis to those with no early relapses; and any early disabling relapses. Analyses were stratified by disease-modifying therapy (DMT) efficacy during follow-up.
Results:
People who experienced non-disabling relapses within 2 years of RRMS diagnosis accumulated more disability than those with no early relapses if they were untreated (n = 285 vs 4717; hazard ratio (HR) = 1.29, 95% confidence interval (CI) = 1.00–1.68) or given platform DMTs (n = 1074 vs 7262; HR = 1.33, 95% CI = 1.15–1.54), but not if given high-efficacy DMTs (n = 572 vs 3534; HR = 0.90, 95% CI = 0.71–1.13) during follow-up. Differences in disability accumulation between those with early non-disabling relapses and those with early disabling relapses were not confirmed statistically.
Conclusion:
This study suggests that early non-disabling relapses are associated with a higher risk of disability accumulation than no early relapses in RRMS. This risk may be mitigated by high-efficacy DMTs. Therefore, non-disabling relapses should be considered when making treatment decisions.
Keywords
Introduction
The prognostic significance of non-disabling relapses in people with relapsing-remitting multiple sclerosis (RRMS) is unclear. However, the European Medicines Agency1,2 restricts the use of certain disease-modifying therapies (DMTs), particularly natalizumab and fingolimod, to only those with disabling relapses. Whether this is justified remains debated 3 but has important implications: early initiation of high-efficacy DMTs can mitigate future disability,4,5 so if early non-disabling relapses predict disability accumulation, disregarding them risks preventable long-term disability. We aimed to determine whether non-disabling relapses early in RRMS predict disability accumulation.
Methods
The MSBase registry was approved by the Melbourne Health Human Research Ethics Committee, and by the local ethics committees in participating centres. Written informed consent was obtained from participants as required.
Longitudinal clinical data from 78,531 patients were extracted from MSBase in May 2022. For inclusion, patients required a diagnosis of clinically-definite RRMS, the MSBase minimal dataset (date of birth, MS centre and dates of MS symptom onset, clinical follow-up, relapses and DMT), a baseline disability score (defined below), and, for the non-disabling relapse group, complete relapse severity information for 2 years from RRMS diagnosis – ‘early MS’.
MSBase classifies relapses as ‘mild’ if they do not affect activities of daily living (ADLs); ‘moderate’ if they affect ADLs; and ‘severe’ if they require hospitalisation. For the purposes of this study, ‘non-disabling’ relapses were those graded as mild by clinicians in MSBase and ‘disabling’ relapses were those graded as moderate or severe, in alignment with treatment guidelines. 6
We compared people with exclusively non-disabling relapses during the 2-year early MS period to: (a) those with no early relapses; and (b) those with at least one early disabling relapse. These relapse severity groups were each stratified by the highest efficacy DMT received: (i) untreated; (ii) only platform DMTs (interferon-beta, glatiramer acetate, dimethyl-fumarate or teriflunomide); 6 and (iii) high-efficacy DMTs (alemtuzumab, anti-CD20 antibodies, cladribine, daclizumab, haematopoietic stem cell transplantation, mitoxantrone, natalizumab, or sphingosine-1-phospate modulators) at any point from RRMS diagnosis to the end of follow-up.
Baseline disability was defined as the first Expanded Disability Status Scale (EDSS) score within the 2-year early MS period when comparing the non-disabling relapse and no relapse group; or the first EDSS score after the early MS period, when comparing with the disabling relapse group, to exclude disability acquired directly from disabling relapses. In case non-disabling relapses also directly caused disability accumulation, a sensitivity analysis was performed comparing the non-disabling relapse and no relapse groups with the baseline as the first EDSS score after the early MS period. Baseline EDSS scores within 60 days after a relapse were excluded. We used mixed-effects Cox models to compare cumulative hazards of a 90-day confirmed disability accumulation event, defined as an increase in EDSS score of ⩾ 1.0 (or ⩾ 1.5 if the baseline EDSS = 0, or 0.5 if the baseline EDSS ⩾ 5.5), adjusted for age, sex, year of baseline EDSS, interval between the first symptom and RRMS diagnosis, EDSS score at RRMS diagnosis and treatment centre (as a random intercept). When comparing the disabling relapse and non-disabling relapse groups, the models were also adjusted for the number of early MS relapses. The Schoenfeld 7 global test was used to detect violation of the proportional hazards assumption. Statistical analysis was performed using R version 4.1.3.
Results
The characteristics of the included patients (Supplemental Figure 1) are outlined in Table 1.
Characteristics of the studied populations.
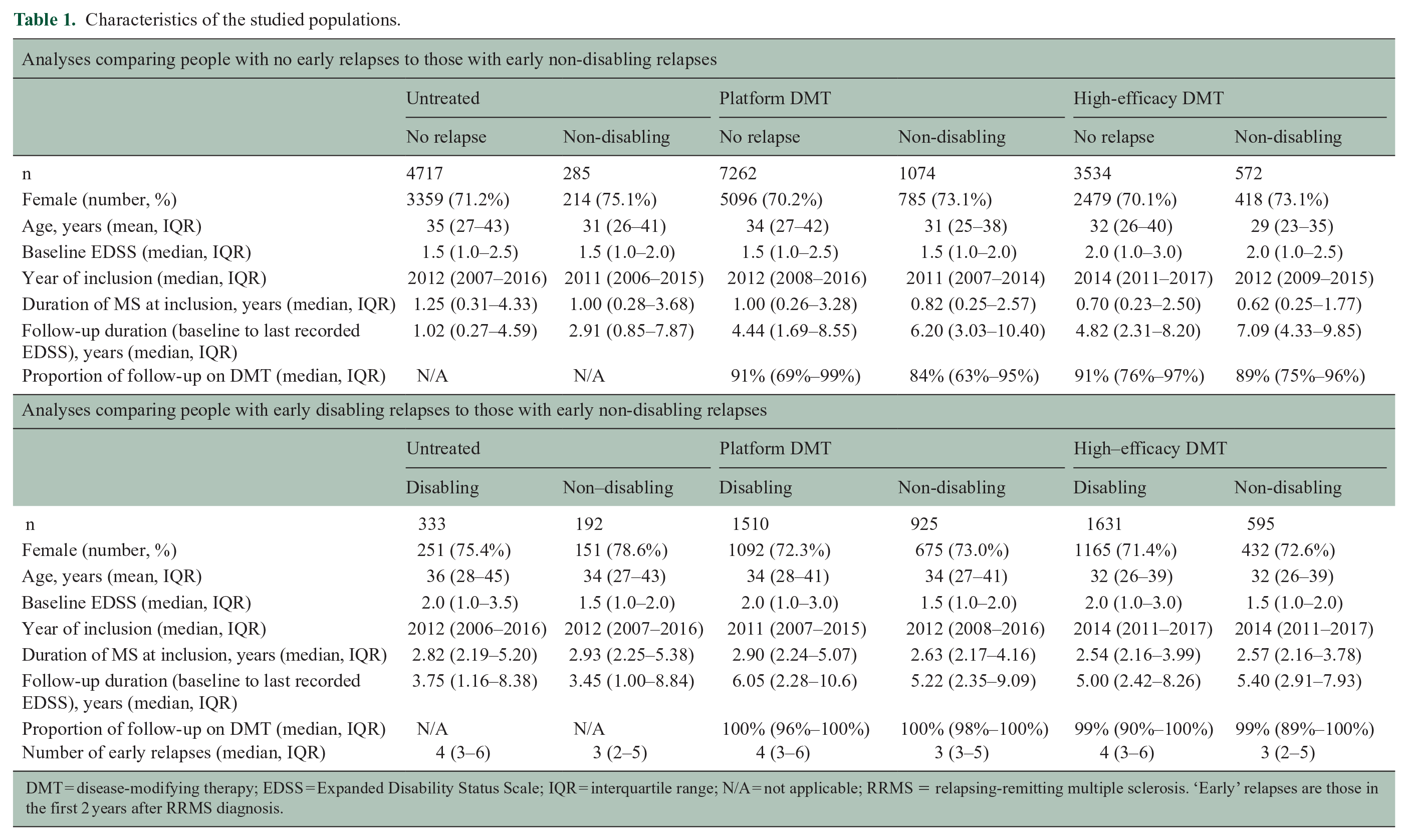
DMT = disease-modifying therapy; EDSS = Expanded Disability Status Scale; IQR = interquartile range; N/A = not applicable; RRMS = relapsing-remitting multiple sclerosis. ‘Early’ relapses are those in the first 2 years after RRMS diagnosis.
People who exclusively experienced non-disabling relapses in the 2-year early MS period accumulated more disability than those with no early relapses if they remained untreated (n = 285 vs 4717; hazard ratio [HR] = 1.29, 95% confidence interval [CI] = 1.00–1.68), or received only platform DMTs (n = 1074 vs 7262; HR = 1.33, 95% CI = 1.15–1.54), but not if they received high-efficacy DMTs (n = 572 vs 3534; HR = 0.90, 95% CI = 0.71–1.13; Figure 1(a)–(c)). These results were similar in a sensitivity analysis with the baseline moved after the early MS period, thus excluding that incomplete recovery from early non-disabling relapses was solely responsible for the observed differences in disability: untreated (n = 192 vs 2449; HR = 1.23; 95% CI = 0.90–1.68); platform DMTs (n = 925 vs 6112; HR = 1.20, 95% CI = 1.03–1.40); high-efficacy DMTs (n = 595 vs 3622; HR = 0.97, 95% CI = 0.78–1.21).
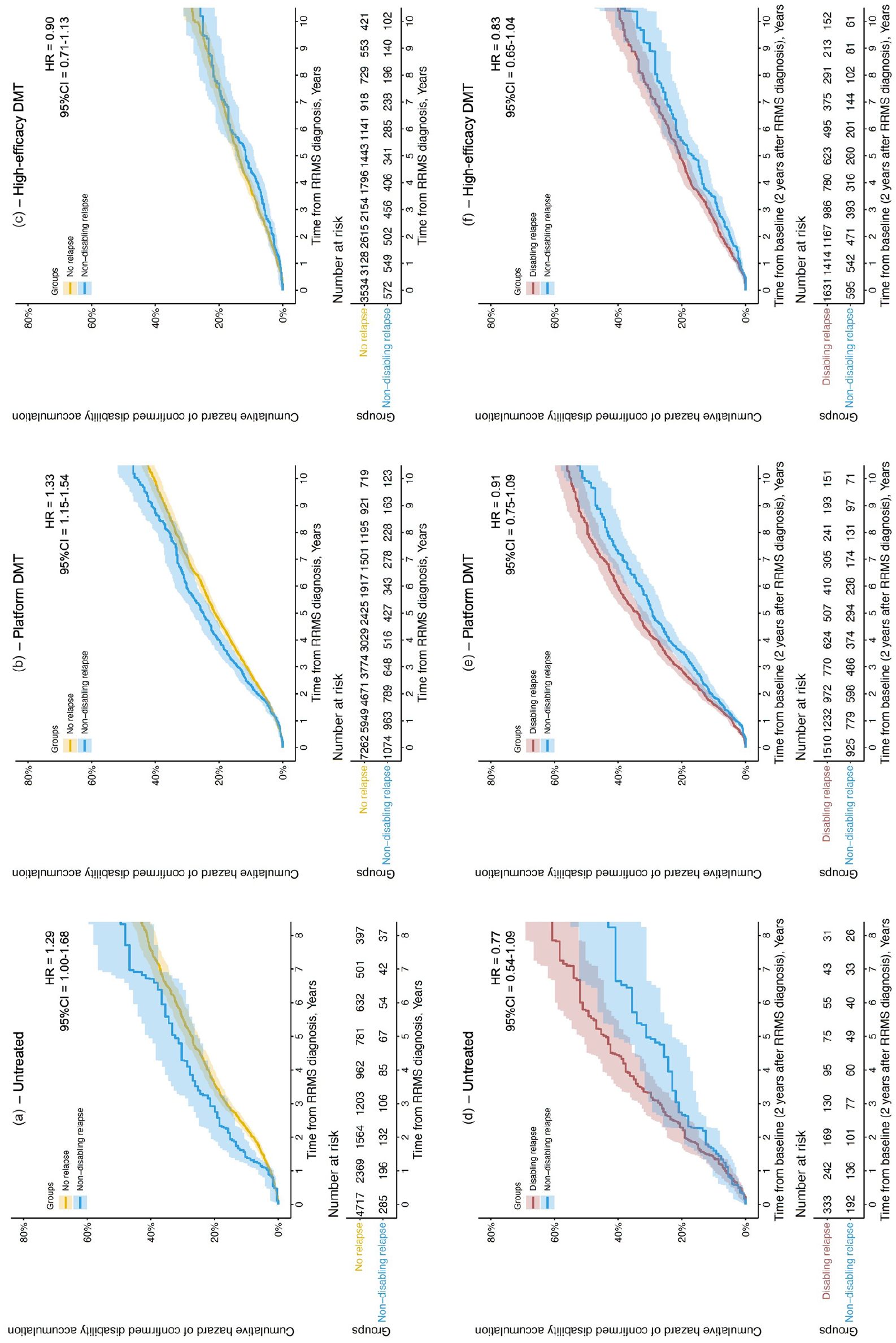
Comparison of the cumulative hazard of a 90-day confirmed disability accumulation event between people with exclusively non-disabling relapses and no relapses (a–c), or exclusively non-disabling relapses and at least one disabling relapse (d–f), in the 2 years after RRMS diagnosis, in people who remained untreated throughout follow-up (a and d), were given only platform DMTs (b and e) or were given high-efficacy DMTs (c and f) at any point during follow-up.
Differences in disability accumulation between people who exclusively experienced non-disabling relapses in the early MS period and those who experienced any disabling relapses in early MS did not reach statistical significance (Figure 1(d)–(f)): untreated (n = 192 vs 333; HR = 0.77; 95% CI = 0.54–1.09); platform DMTs (n = 925 vs 1510; HR = 0.91, 95% CI = 0.75–1.09); high-efficacy DMTs (n = 595 vs 1631; HR = 0.83, 95% CI = 0.65–1.04).
Discussion
In this international observational study, people with RRMS who experienced early non-disabling relapses had a higher risk of disability accumulation than those with no early relapses, if they were untreated or received only platform DMTs during follow-up. However, this association was not observed in people who received high-efficacy DMTs. This suggests, contrary to current guidance,1,2 that non-disabling relapses should be considered in decisions to initiate or escalate treatment, including with high-efficacy therapies.
We did not confirm statistically whether distinguishing early relapse severity has prognostic significance, but the power of these analyses was limited by smaller numbers. There was a non-significant trend towards more disability accumulation with early disabling relapses over early non-disabling relapses, which was mitigated with DMTs. This might be explained by previous observations that relapse phenotypes tend to recur, 8 and incomplete recovery from the first relapse, which is more common for disabling relapses, predicts incomplete recovery from subsequent relapses. 9
Limitations
Relapse severity was non-standardised. However, the three-category classification is defined by the MSBase Study Protocol and reflects current DMT-prescribing restrictions 6 as applied in real-world clinical practice. By including treatment centre in our models, we mitigated variation among centres in relapse severity classification. Data were missing on the severity of a large proportion of early relapses, which may have limited both our power to detect differences between disabling relapses and non-disabling relapses and the generalisability of our conclusions. We also did not perform a direct comparison between matched groups of patients treated with different DMT efficacies following a non-disabling relapse. In view of these limitations, further studies are required to confirm the futility of making treatment decisions based on relapse severity.
The follow-up duration in the untreated populations was short (median 1.02–3.75 years). However, a similar risk of disability was observed in patients with non-disabling relapses treated with platform DMTs over a longer follow-up (median 4.44–6.20 years).
On-treatment relapses may be associated with worse outcomes than off-treatment relapses, 10 but we did not explore this here. We also did not explore the prognostic value of radiological disease activity.
Conclusion
This study suggests that people with early non-disabling relapses have a higher risk of disability accumulation than those with no early relapses and that this risk might be mitigated by high-efficacy DMTs. Therefore, even non-disabling relapses should be considered when making treatment decisions.
Supplemental Material
sj-docx-1-msj-10.1177_13524585231151951 – Supplemental material for Early non-disabling relapses are important predictors of disability accumulation in people with relapsing-remitting multiple sclerosis
Supplemental material, sj-docx-1-msj-10.1177_13524585231151951 for Early non-disabling relapses are important predictors of disability accumulation in people with relapsing-remitting multiple sclerosis by Cyrus Daruwalla, Vahid Shaygannejad, Serkan Ozakbas, Eva Kubala Havrdova, Dana Horakova, Raed Alroughani, Cavit Boz, Francesco Patti, Marco Onofrj, Alessandra Lugaresi, Sara Eichau, Marc Girard, Alexandre Prat, Pierre Duquette, Bassem Yamout, Samia J Khoury, Seyed Aidin Sajedi, Recai Turkoglu, Ayse Altintas, Olga Skibina, Katherine Buzzard, Pierre Grammond, Rana Karabudak, Anneke van der Walt, Helmut Butzkueven, Davide Maimone, Jeannette Lechner-Scott, Aysun Soysal, Nevin John, Julie Prevost, Daniele Spitaleri, Cristina Ramo-Tello, Oliver Gerlach, Gerardo Iuliano, Matteo Foschi, Radek Ampapa, Vincent van Pesch, Michael Barnett, Nevin Shalaby, Marie D’hooghe, Jens Kuhle, Maria Jose Sa, Marzena Fabis-Pedrini, Allan Kermode, Saloua Mrabet, Riadh Gouider, Suzanne Hodgkinson, Guy Laureys, Liesbeth Van Hijfte, Richard Macdonell, Celia Oreja-Guevara, Edgardo Cristiano, Pamela McCombe, Jose Luis Sanchez-Menoyo, Bhim Singhal, Yolanda Blanco, Stella Hughes, Justin Garber, Claudio Solaro, Chris McGuigan, Bruce Taylor, Koen de Gans, Mario Habek, Abdullah Al-Asmi, Simu Mihaela, Tamara Castillo Triviño, Talal Al-Harbi, Juan Ignacio Rojas, Orla Gray, Dheeraj Khurana, Bart Van Wijmeersch, Nikolaos Grigoriadis, Jihad Inshasi, Jiwon Oh, Eduardo Aguera-Morales, Yara Fragoso, Fraser Moore, Cameron Shaw, Seyed Mohammad Baghbanian, Neil Shuey, Barbara Willekens, Todd A Hardy, Danny Decoo, Angel Perez sempere, Deborah Field, Ray Wynford-Thomas, Nick G Cunniffe, Izanne Roos, Charles B Malpas, Alasdair J Coles, Tomas Kalincik and J William L Brown in Multiple Sclerosis Journal
Footnotes
Acknowledgements
MSBase study group contributors included the following: from Department of Medical and Surgical Sciences and Advanced Technologies, GF Ingrassia, Catania, Italy, Dr Clara Chisari, Dr Emanuele D’Amico, Dr Lo Fermo Salvatore; from University G. d’Annunzio, Chieti, Italy, Dr Giovanna De Luca, Dr Valeria Di Tommaso, Dr Daniela Travaglini, Dr Erika Pietrolongo, Dr Maria di Ioia, Dr Deborah Farina; from CHUM and Universite de Montreal, Montreal, Canada, Dr Catherine Larochelle, from MS Centre, Department of Neurology, Royal Melbourne Hospital, Melbourne, Australia, Dr Mark Marriott, Dr Trevor Kilpatrick, Dr John King, Dr Katherine Buzzard, Dr Ai-Lan Nguyen, Dr Chris Dwyer, Dr Mastura Monif, Ms Lisa Taylor, Ms Josephine Baker; from Academic MS Center Zuyderland, Department of Neurology, Zuyderland Medical Center, Sittard-Geleen, The Netherlands, Dr Raymond Hupperts; from Azienda Sanitaria Unica Regionale Marche–AV3, Macerata, Italy, Dr Matteo Diamanti; from Centro de Esclerosis Múltiple de Buenos Aires (CEMBA), Buenos Aires, Argentina, Dr Juan Ingacio Rojas, from Groene Hart Ziekenhuis, Gouda, Netherlands, Dr Freek Verheul; and from Hospital Universitario Donostia and IIS Biodonostia, San Sebastián, Spain, Dr Javier Olascoaga.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: E.K.H. received honoraria/research support from Biogen, Merck Serono, Novartis, Roche, and Teva; has been member of advisory boards for Actelion, Biogen, Celgene, Merck Serono, Novartis, and Sanofi Genzyme. She was also supported by the Charles University: Cooperatio Program in neuroscience. D.H. received compensation for travel, speaker honoraria and consultant fees from Biogen, Novartis, Merck Healthcare KGaA (Darmstadt, Germany), Bayer, Sanofi, Roche, and Teva, as well as support for research activities from Biogen. She was also supported by the Charles University: Cooperatio Program in neuroscience. R.A. received honoraria as a speaker and for serving on scientific advisory boards from Bayer, Biogen, GSK, Merck, Novartis, Roche and Sanofi-Genzyme. C.B. received conference travel support from Biogen, Novartis, Bayer-Schering, Merck and Teva; has participated in clinical trials by Sanofi Aventis, Roche and Novartis.
F.P. received speaker honoraria and advisory board fees from Almirall, Bayer, Biogen, Celgene, Merck, Novartis, Roche, Sanofi-Genzyme and TEVA. He received research funding from Biogen, Merck, FISM (Fondazione Italiana Sclerosi Multipla), Reload Onlus Association and University of Catania.
A.L. has received personal compensation for consulting, serving on a scientific advisory board, speaking or other activities from Biogen, Merck Serono, Mylan, Novartis, Roche, Sanofi/Genzyme and, Teva, and Bristol Myers Squibb. Her institutions have received research grants from Novartis and Sanofi/Genzyme.
S.E. received speaker honoraria and consultant fees from Biogen Idec, Novartis, Merck, Bayer, Sanofi Genzyme, Roche and Teva.
M.G. received consulting fees from Teva Canada Innovation, Biogen, Novartis and Genzyme Sanofi; lecture payments from Teva Canada Innovation, Novartis and EMD . He has also received a research grant from Canadian Institutes of Health Research.
S.J.K. received compensation for scientific advisory board activity from Merck and Roche.
A.A. received speaker honoraria from Merck, Alexion; received travel and registration grants from Merck.
K.B. received honoraria and consulting fees from Biogen, Teva, Novartis, Genzyme-Sanofi, Roche, Merck, CSL and Grifols.
P.G. has served in advisory boards for Novartis, EMD Serono, Roche, Biogen Idec, Sanofi Genzyme, Pendopharm and has received grant support from Genzyme and Roche, has received research grants for his institution from Biogen Idec, Sanofi Genzyme, EMD Serono.
A.v.d.W. served on advisory boards and receives unrestricted research grants from Novartis, Biogen, Merck and Roche She has received speaker’s honoraria and travel support from Novartis, Roche, and Merck. She receives grant support from the National Health and Medical Research Council of Australia and MS Research Australia.
H.B. received institutional (Monash University) funding from Biogen, F. Hoffmann-La Roche Ltd, Merck, Alexion, CSL, and Novartis; has carried out contracted research for Novartis, Merck, F. Hoffmann-La Roche Ltd and Biogen; has taken part in speakers’ bureaus for Biogen, Genzyme, UCB, Novartis, F. Hoffmann-La Roche Ltd and Merck; has received personal compensation from Oxford Health Policy Forum for the Brain Health Steering Committee.
D.M. received speaker honoraria for Advisory Board and travel grants from Almirall, Biogen, Merck, Novartis, Roche, Sanofi-Genzyme, and Teva.
J.L-.S. received travel compensation from Novartis, Biogen, Roche and Merck. Her institution receives the honoraria for talks and advisory board commitment as well as research grants from Biogen, Merck, Roche, TEVA and Novartis.
N.J. is a local principal investigator on commercial studies funded by Novartis, Biogen, Amicus and Sanofi
J.P. accepted travel compensation from Novartis, Biogen, Genzyme, Teva, and speaking honoraria from Biogen, Novartis, Genzyme and Teva.
D.S. received honoraria as a consultant on scientific advisory boards by Bayer-Schering, Novartis and Sanofi-Aventis and compensation for travel from Novartis, Biogen, Sanofi Aventis, Teva and Merck.
C.R-.T. received research funding, compensation for travel or speaker honoraria from Biogen, Novartis, Genzyme and Almirall.
G.I. had travel/accommodations/meeting expenses funded by Bayer Schering, Biogen, Merck, Novartis, Sanofi Aventis, and Teva.
R.A. received conference travel support from Novartis, Teva, Biogen, Bayer and Merck and has participated in a clinical trial by Biogen, Novartis, Teva and Actelion.
V.v.P. received travel grants from Merck Healthcare KGaA (Darmstadt, Germany), Biogen, Sanofi, Bristol Meyer Squibb, Almirall and Roche. His institution has received research grants and consultancy fees from Roche, Biogen, Sanofi, Merck Healthcare KGaA (Darmstadt, Germany), Bristol Meyer Squibb, Janssen, Almirall and Novartis Pharma
M.B. served on scientific advisory boards for Biogen, Novartis and Genzyme and has received conference travel support from Biogen and Novartis. He serves on steering committees for trials conducted by Novartis. His institution has received research support from Biogen, Merck and Novartis.
M.D. received consultancy and advisory board fees from Roche, Sanofi-Genzyme, Biogen, Merck-Serono, Bayer-Schering, Novartis and Allergan; received congress support from Biogen, Merck-Serono, Teva and Roche. She has also received research support from Novartis, Biogen, Roche, FWO (Research Foundation Flanders) and Fonds D.V. (Ligue Nationale Belge de la Sclerose en Plaques, Fondation Roi Baudouin).
J.K. received speaker fees, research support, travel support, and/or served on advisory boards by Swiss MS Society, Swiss National Research Foundation (320030_189140/1), University of Basel, Progressive MS Alliance, Bayer, Biogen, Bristol Myers Squibb, Celgene, Merck, Novartis, Octave Bioscience, Roche, Sanofi.
M.J.S. received consulting fees, speaker honoraria, and/or travel expenses for scientific meetings from Alexion, Bayer Healthcare, Biogen, Bristol Myers Squibb, Celgene, Janssen, Merck-Serono, Novartis, Roche, Sanofi and Teva.
M.F-.P. received travel compensation from Merck
A.K. received speaker honoraria and scientific advisory board fees from Bayer, BioCSL, Biogen, Genzyme, Innate Immunotherapeutics, Merck, Novartis, Sanofi, Sanofi-Aventis, and Teva.
S.M. has received a MENACTRIMS clinical fellowship grant (2020).
S.H. received honoraria and consulting fees from Novartis, Bayer Schering and Sanofi, and travel grants from Novartis, Biogen Idec and Bayer Schering.
G.L. received travel and/or consultancy compensation from Sanofi-Genzyme, Roche, Teva, Merck, Novartis, Celgene, Biogen.
C.O-.G. received honoraria as consultant on scientific advisory boards from Biogen, Celgene, Merck, Novartis, Roche, Sanofi-Genzyme and TEVA.
E.C. received honoraria as consultant on scientific advisory boards by Biogen, Bayer-Schering, Merck, Genzyme and Novartis; has participated in clinical trials/other research projects by Merck, Roche and Novartis.
P.M. received speakers fees and travel grants from Novartis, Biogen, T’évalua, Sanofi and Bayer Schering and received honoraria and consulting fees from Novartis, Bayer Schering and Sanofi.
J.L.S-.M. accepted travel compensation from Novartis, Merck and Biogen, speaking honoraria from Biogen, Novartis, Sanofi, Merck, Almirall, Bayer and Teva and has participated in clinical trials by Biogen, Merck and Roche
B.S. received consultancy honoraria and compensation for travel from Biogen and Merck.
S.H. has received unrestricted educational grants or speaking honoraria from Biogen, Merck Serono, Novartis, Roche and Sanofi Genzyme.
Y.B. received speaker honoraria from Merck, Biogen, Brystol, Novartis and Sanofi
C.S. served on scientific advisory boards for Merck, Genzyme, Almirall, and Biogen; received honoraria and travel grants from Sanofi Aventis, Novartis, Biogen, Merck, Genzyme and Teva.
B.T. received funding for travel and speaker honoraria from Bayer Schering Pharma, CSL Australia, Biogen and Novartis, and has served on advisory boards for Biogen, Novartis, Roche and CSL Australia.
M.H. participated as a clinical investigator and/or received consultation and/or speaker fees from Biogen, Sanofi Genzyme, Merck, Bayer, Novartis, Pliva/Teva, Roche, Alvogen, Actelion, Alexion Pharmaceuticals and TG Pharmaceuticals
S.M. received speaker honoraria, advisory board fees and travel grants from Abbvie, Biogen, Bristol Meyrs Squibb, Teva, Merck, Roche, Ipsen, Sanofi-Genzyme Novartis, Boehringer Stada
T.C.T. received speaking/consulting fees and/or travel funding from Almirall, Biogen, Bristol Myers Squibb, Merck, Novartis, Roche, Sanofi-Genzyme and Teva.
O.G. received honoraria as consultant on scientific advisory boards for Genzyme, Biogen, Merck, Roche and Novartis; has received travel grants from Biogen, Merck, Roche and Novartis; has participated in clinical trials by Biogen and Merck.
B.V.W. received research and travel grants, honoraria for MS-Expert advisor and Speaker fees from Almirall, Biogen, BMS, Janssen, Sanofi, Merck, Novartis, Roche and Teva.
N.G. received honoraria and travel support, Consultancy fees, Lecture fees from Biogen Idec, Biologix, Novartis, TEVA, Bayer, Merck Serono, Genesis Pharma, Sanofi – Genzyme, ROCHE, ELPEN. Research grants from Biogen Idec, Novartis, TEVA, Merck Serono, Genesis Pharma, Sanofi – Genzyme, ROCHE
J.O. has received research funding from the MS Society of Canada, National MS Society, Brain Canada, Biogen, Roche, EMD Serono (an affiliate of Merck KGaA); and personal compensation for consulting or speaking from Alexion, Biogen, Celgene (BMS), EMD Serono (an affiliate of Merck KGaA), Novartis, Roche, and Sanofi-Genzyme.
Y.F. received honoraria as a consultant on scientific advisory boards by Novartis, Teva, Roche and Sanofi-Aventis and compensation for travel from Novartis, Biogen, Sanofi Aventis, Teva, Roche and Merck.
F.M. participated in clinical trials sponsored by EMD Serono and Novartis.
C.S. received travel assistance from Biogen and Novartis.
N.S. received travel compensation from Bayer Schering, Novartis, and Biogen Idec.
B.W. received honoraria for acting as a member of Scientific Advisory Boards for Almirall, Biogen, Celgene/BMS, Merck Serono, Novartis, Roche, Sanofi-Genzyme and speaker honoraria and travel support from Biogen, Merck Serono, Novartis, Roche, Sanofi-Genzyme; research and/or patient support grants from Roche, Biogen, Merck-Serono, Sanofi-Genzyme; research support from FWO. Honoraria and grants were paid to UZA/UZA Foundation.
T.A.H. has received speaking fees or received honoraria for serving on advisory boards for Biogen, Merck, Teva, Novartis, Roche, Bristol Myers Squibb and Sanofi.
D.D. received compensation for travel, speaker honoraria and consultant fees from Biogen, Novartis, Merck KGaA, Bayer, Sanofi/Genzyme, Roche and Teva, as well as support for research activities from Biogen, Novartis, Merck, Sanofi, Roche & Teva
R.W-.T.’s clinical research fellow grant is co-funded by Novartis.
I.R. served on scientific advisory boards, received conference travel support and/or speaker honoraria from Roche, Novartis, Merck and Biogen. Izanne Roos receives research support from Multiple Sclerosis Australia and the Trish Multiple Sclerosis Research Foundation.
C.M. has received conference travel support from Merck, Novartis, and Biogen. He has received research support from the National Health and Medical Research Council, Multiple Sclerosis Research Australia, The University of Melbourne, The Royal Melbourne Hospital Neuroscience Foundation, and Dementia Australia.
T.K. served on scientific advisory boards for BMS, Roche, Janssen, Sanofi Genzyme, Novartis, Merck and Biogen, steering committee for Brain Atrophy Initiative by Sanofi Genzyme, received conference travel support and/or speaker honoraria from WebMD Global, Eisai, Novartis, Biogen, Sanofi-Genzyme, Teva, BioCSL and Merck and received research or educational event support from Biogen, Novartis, Genzyme, Roche, Celgene and Merck.
J.W.L.B. received speaking honoraria and advisory board fees from Biogen, Novartis, Intesso and The Corpus.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by National Health and Medical Research Council of Australia (fellowship nos.1140766 and 1080518, project grant nos. 1129189 and 1083539), the University of Melbourne (Faculty of Medicine, Dentistry and Health Sciences research fellowship), National Institute for Health and Care Research (UK) Advanced Fellowship (grant no. 301728; recipient JWLB) and Academic Clinical Fellowship (grant no. EAN/ACA-006/7488627/C; recipient CD). The MSBase Foundation is a not-for-profit organization that receives support from Roche, Merck, Biogen, Novartis, Bayer Schering, Sanofi Genzyme, and Teva. Role of the Funder/Sponsor: The National Health and Medical Research Council of Australia, the University of Melbourne and the National Institute for Health and Care Research (UK) had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
ORCID iDs
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.