
Abstract
Background:
Accumulating evidence supports the efficacy of administering natalizumab (NZ) with extended-interval dosing (EID) in patients with relapsing-remitting multiple sclerosis (RRMS).
Objectives:
We switched NZ dosing from 4-week to 6-week intervals in patients with RRMS, and investigated the effect on serum neurofilament light chain (sNfL) concentrations.
Methods:
We included two cohorts of patients with RRMS treated with NZ: one received the standard-interval dosing (4 weeks) at baseline, and were switched to 6-week intervals (EID4–6, N = 45). The other cohort received EID (5- or 6-week intervals) both at baseline and during follow-up (EID5/6, N = 25). Serum samples were collected in the EID4–6 cohort at every NZ infusion, for 12 months. The primary outcome was the change in sNfL concentrations after switching to EID.
Results:
The baseline mean sNfL concentration in the EID4–6 cohort was 10.5 ng/L (standard deviation (SD) = 6.1), and it remained unchanged at 12 months. Moreover, individual sNfL concentrations did not change significantly after extending the NZ dosing intervals. In addition, the EID4–6 and EID5/6 cohorts had similar baseline sNfL concentrations.
Conclusion:
We concluded that extending the NZ dosing interval did not increase axonal damage, as determined with sNfL, in patients with RRMS.
Introduction
Natalizumab (NZ) is a monoclonal antibody used for treating patients with relapsing-remitting multiple sclerosis (RRMS). 1 NZ is administered according to a standard dosing schedule of 300 mg every 4 weeks (standard-interval dosing; SID). When NZ binds to α4 integrin on the surface of leukocytes, it prevents leukocyte migration from the blood into the central nervous system (CNS). NZ effectively reduces disease activity in RRMS, and it is well tolerated with a few adverse effects. 2 The main drawback of NZ is that it increases the risk of progressive multifocal leukoencephalopathy (PML), 3 a John Cunningham (JC virus) virus infection that often gives rise to severe impairment and is lethal in 24% of patients treated with NZ. 4 In non-randomized observational studies, extended-interval dosing (EID) was associated with a significantly lower risk of developing PML, compared to SID, 5 but it had similar therapeutic efficacy.6–9 Very recently, therapeutic efficacy has also been demonstrated to be maintained in EID with NZ in a randomized controlled study. 10
Previously, when switching NZ dosing from SID to EID, disease activity and progression was generally monitored with conventional cerebral magnetic resonance imaging (MRI) and clinical evaluations.6,7,9,11 However, current evidence has suggested that signs of inflammatory activity and neurodegeneration may escape detection with conventional monitoring.12–14
The most promising soluble biomarker in MS is neurofilament light (NfL), 15 a marker of axonal damage that can be determined in cerebrospinal fluid (CSF) 16 as well as in blood.17,18 There is accumulating evidence that NfL is a reliable biomarker of disease activity in RRMS,19,20 that may also reflect therapeutic efficacy. 21 Consequently, NfL has served as an additional outcome measure in clinical trials. 22
This study aimed to determine whether switching NZ treatment intervals from SID to EID might affect serum neurofilament light chain (sNfL) concentrations in patients with RRMS. To reduce potential effects of other factors on sNfL levels, we selected patients who lacked signs of disease activity in clinical and MRI examinations.
Material and methods
Study design and patients
This prospective observational single-center study was conducted for 12 months at the MS center, Sahlgrenska University Hospital in Gothenburg, Sweden. Eligible patients had RRMS, fulfilled the 2017 McDonald criteria, 23 and had been receiving 300 mg NZ (Tysabri®, Biogen, Cambridge, MA, USA) intravenously, every 4, 5, or 6 weeks, for at least 1 year. They should not have any relapse or new or enlarging lesions on MRI within 6 months prior to baseline. After signing informed consent forms, patients were consecutively enrolled in the study. The first patients were included on the 1st of Oct 2019 and the last follow-up visit was on the 1st of June 2021. The inclusion process is illustrated in Figure 1.
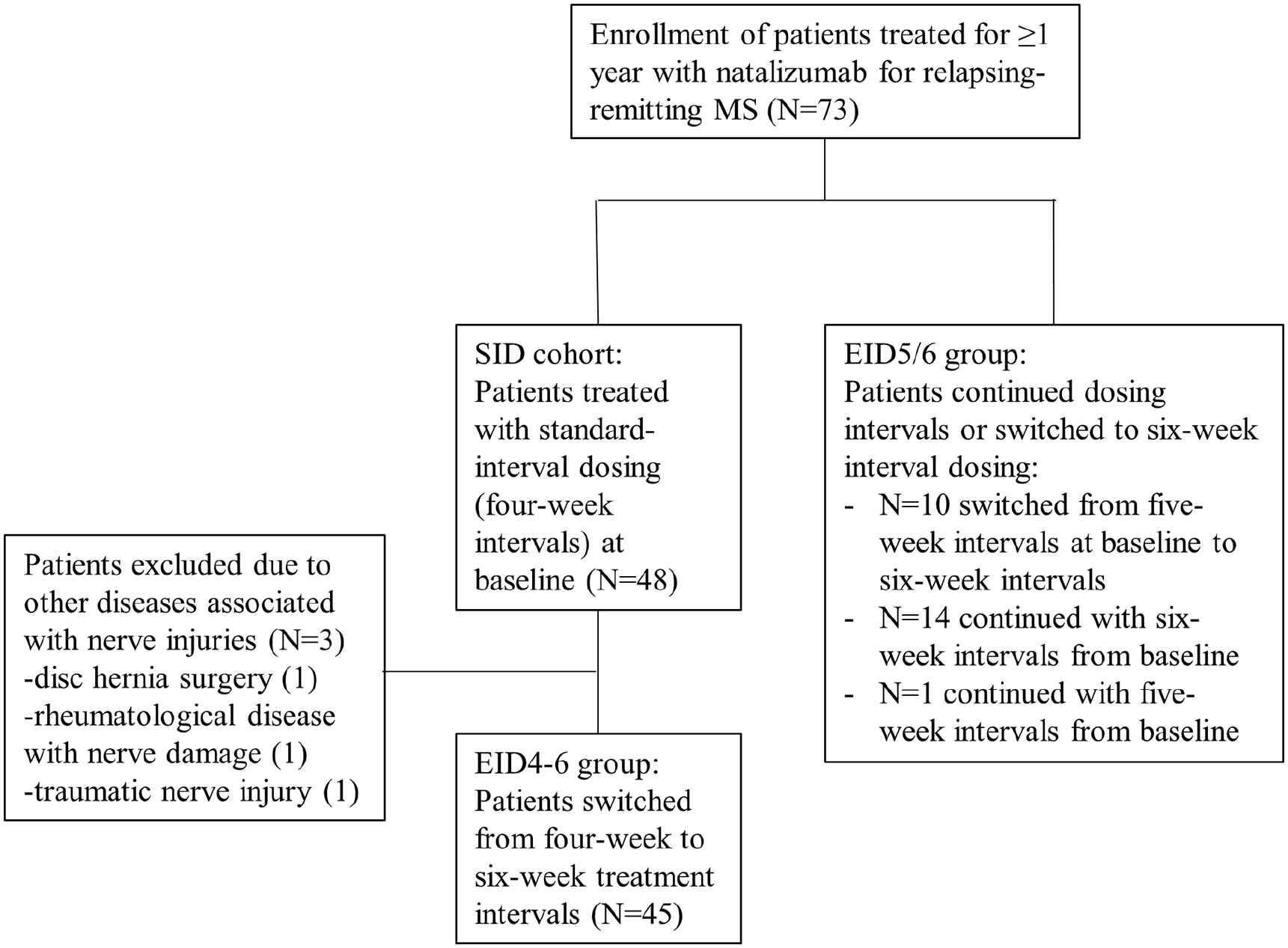
Flow chart of patient selection and treatment allocation.
Procedures
The study participants were divided into two cohorts, based on the NZ infusion interval; one cohort had received NZ at 4-week intervals prior to baseline, and they were switched to receive EID at 6-week intervals (EID4–6). The other group had received EID at 5- or 6-week intervals at baseline, and continued extended dosing (EID5/6). Except for one patient, the patients in EID5/6 who received EID at 5-week intervals at baseline switched to 6-week intervals.
In the EID4–6 cohort, peripheral blood was drawn at 4 weeks prior to baseline, at baseline (week zero), and then every 6 weeks, up to 48 weeks. In the EID5/6 cohort, blood was drawn at baseline (week zero), at 5/6 weeks, and at 12 weeks. In both study cohorts, MRI scan was performed at baseline, at 24 weeks, and at 48 weeks, and clinical examination with Expanded Disability Status Scale (EDSS) 24 scoring were performed at baseline and at 48 weeks (Figure 2).
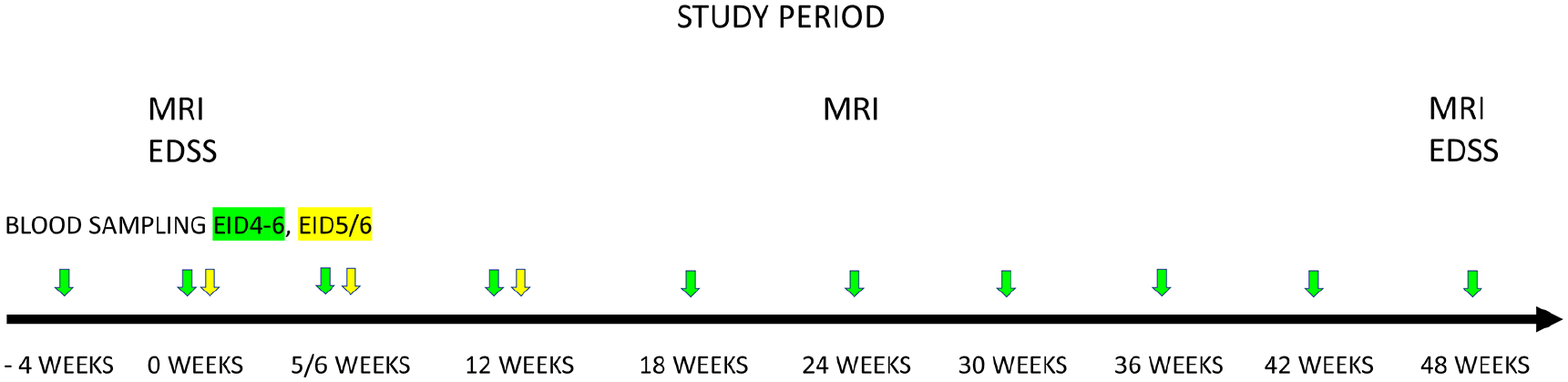
Study design for testing the effects of extended natalizumab dosing in patients with relapsing-remitting MS. Patients in the EID4–6 treatment group (green) switched from standard (4-week) to extended (6-week) dosing intervals; patients in the EID5/6 group (yellow) remained on extended dosing intervals (i.e. 5- or 6-week intervals). Blood samples were drawn (color-coded arrows) to analyze serum neurofilament light chain concentrations. Patients underwent conventional monitoring with MRI and EDSS at the indicated times.
Baseline demographic and clinical characteristics were retrieved from the Swedish MS Registry (SMSreg, http://www.msreg.net) and from electronic patient records. A relapse was defined as the appearance of new or worsening neurological symptoms compatible with MS that lasted more than 24 hours in the absence of any factor that could cause worsening of symptoms (i.e. a pseudo relapse). Disability was assessed at clinical visits or via telephone due to the COVID-19 pandemic, and scored with EDSS or telephone-EDSS. 25 Significant disability progression was defined as an increase of 1.5 points from a baseline EDSS score of 0, an increase of 1 point from a baseline EDSS score of 1–5.5, and an increase of 0.5 point from a baseline EDSS score >5.5. The MRI protocol included the brain and cervical spinal cord with T1-weighted images, T1-weighted images with gadolinium contrast, T2-weighted images, fluid-attenuated inversion recovery (FLAIR) images, and diffusion-weighted imaging (DWI), performed according to the Swedish guidelines. 26 No evidence of disease activity (NEDA-3) was defined as a lack of relapse, no new or enlarging lesions detected on MRIs, and no significant progression during the study period. 27
Intravenous peripheral blood samples were obtained prior to NZ infusion. Samples were collected in three pairs of 5-mL serum-gel and plasma containers. Serum samples were maintained at room temperature for 30 minutes to allow complete clotting. The samples were spun at 2000g for 10 minutes, then aliquoted in 1-mL portions and frozen directly at −80°C.
All NfL analyses were performed by board-certified laboratory technicians who were blinded to clinical data. To minimize variation, baseline and follow-up samples were analyzed side-by-side on each assay plate using one batch of reagents. In addition, samples from healthy controls were randomly analyzed in each assay plate. All analyses were performed at room temperature. Serum NfL concentration was measured using the Simoa® NF-light™ Advantage Kit on an HD-X Analyzer (Quanterix, Billerica, MA, USA). Briefly, the samples, including internal quality control samples, and calibrator stock were removed from storage and allowed to thaw at room temperature. The RGP reagent was shaken for 30 minutes at 800 r/min and heated to 30°C. The calibrators, samples, and QCs were vortexed for 30 seconds at 2000 r/min. The internal calibrators, samples, and QCs were additionally centrifuged for 10 minutes at 4000g. Calibrators, samples, and QCs were added to the plate and covered with sealing tape. Reagents, samples, and calibrators were run in the HD-1 Analyzer using a 4× dilution. The intra-assay and inter-assay coefficients of variation were 10%.
Standard protocol approvals, registrations, and patient consents
This study was conducted in accordance with the Declaration of Helsinki and the International Good Clinical Practice guideline. The study was approved by the Regional Committee for Medical Research Ethics, Gothenburg (EPN-460-13) and the Swedish Ethical Review Agency (DNR 2020-04900). Written informed consent was obtained from all participating patients.
Statistical analyses
Statistical analyses were performed with SAS 9.4 (SAS Institute Inc., Cary, NC, USA), SPSS version 23 (IBMCorp., Armonk, NY, USA), and GraphPad Prism 9.3 (GraphPad Software, San Diego, CA, USA). For continuous variables, comparisons between groups were performed with Fisher’s Non-Parametric Permutation Test. 28 For matched pairs, comparisons within groups were performed with Fischer’s Non-Parametric Permutation test. Analysis of covariance (ANCOVA) was performed to adjust for age. The Wilcoxon signed-rank test was performed to evaluate differences between sNfL and plasma neurofilament light chain (pNfL) and changes in EDSS from baseline to 12 months. Pearson’s correlation coefficients were used to evaluate correlations between sNfL and age or body mass index (BMI). Pearson’s correlation analysis and the Shrout-Fleiss reliability random test 29 were performed to compare sNfL and pNfL. The Mersenne Twister was used for random number generation.
Results
The study included 73 patients with RRMS; 48 had been treated with SID and 25 had been treated with EID. Three patients in the EID4–6 cohort were excluded due to concomitant conditions with neurological injuries (Figure 1). The demographic and clinical characteristics of the study cohorts are presented in Table 1.
Baseline demographic and clinical characteristics of patients with RRMS.

RRMS: relapsing-remitting multiple sclerosis; EID: extended-interval dosing; EID4–6: patients switched from treatment at 4-week intervals to treatment at 6-week intervals; EID5/6: patients treated at 5- or 6-week intervals; BMI: body mass index; EDSS: Expanded Disability Status Scale; NZ: natalizumab; MS: multiple sclerosis; DMT: disease-modifying treatment; SID: standard-interval dosing; JC virus: John Cunningham virus.
Values are the mean (range), unless indicated otherwise.
One patient in the EID5/6 cohort experienced a relapse and a new non-enhancing lesion was detected on MRI. However, none of the other patients exhibited clinical or MRI signs of disease activity. In the EID4–6 cohort, no significant change was observed between the mean EDSS values at baseline and at 48 weeks (p = 0.68). Although three patients converted from RRMS to secondary progressive MS during follow-up, only one showed a significant increase in the EDSS. Of the other two patients, one showed an increase of 0.5 points in the EDSS, and the other showed no change in the EDSS, but experienced a progressive reduction in walking distance. Accordingly, overall, NEDA-3 was achieved in 66/70 patients (94%).
sNfL concentrations compared between SID and EID treatment groups
In the EID4–6 cohort, the mean sNfL concentration at baseline (week zero) was 10.5 ng/L (standard deviation (SD) = 6.1) (i.e. before switching from the 4-week to the 6-week dosing interval). We compared changes in mean sNfL concentrations between all samples, but also between baseline and the early period (weeks 6–18), and between baseline and the entire study period (weeks 6–48). Serum NfL concentrations remained stable throughout the 48-week study period with no significant change in the mean sNfL at any time-point (Figure 3).
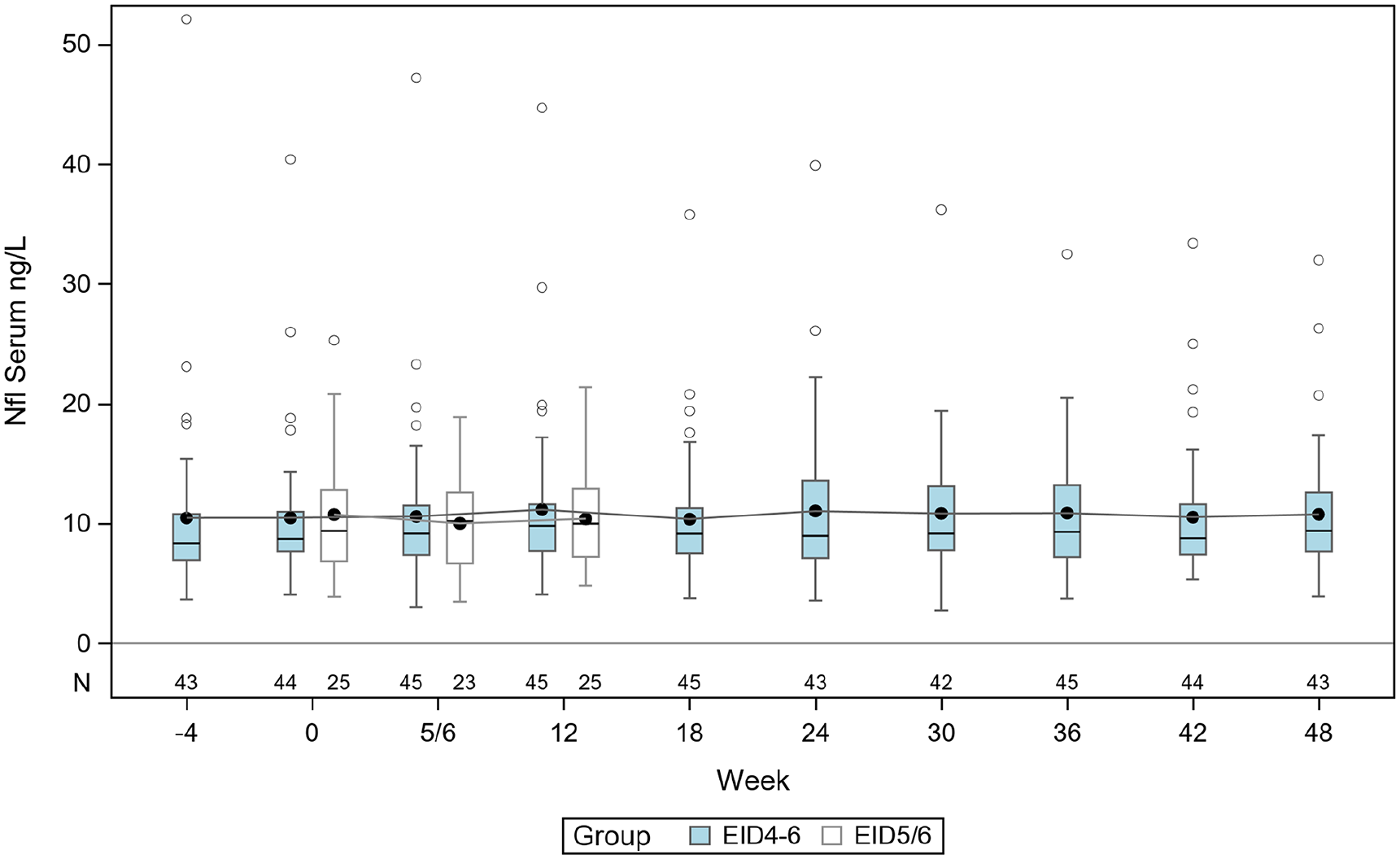
Serum NfL levels in ng/L in group EID4–6 (blue) and EID5/6 (white) where line in box is median and marker shows the mean. The boxes indicate the interquartile range and the vertically extending lines are minimum and maximum except for individual samples indicated as circles.
In the EID5/6 cohort, the mean sNfL concentration at baseline was 10.8 ng/L (SD = 5.3) and for all samples 10.3 ng/L (SD = 4.2), which was of similar level as the mean sNfL in the EID4–6 cohort (10.5 ng/L; SD = 6.1) before extending the NZ dosing interval (baseline). There was no statistically significant difference in the baseline mean sNfL between EID4–6 and EID5/6 (−0.28 ng/L, 95% confidence interval (CI) = −2.97–2.70). Hence, we found no significant increase of sNfL in NZ-treated patients with EID compared with SID. Furthermore, the sNfL concentrations did not change significantly in the patient who experienced a relapse and a non-enhancing new lesion or in patients who converted to secondary progressive MS.
Inter- and intra-individual variation of sNfL in NZ-treated RRMS
In the EID4-6 cohort, we investigated the inter- and intra-individual variation of sNfL in order to evaluate sNfL as a biomarker for individual patients. The mean sNfL (all samples) was 10.8 ng/L (SD 5.9), and the median sNfL 8.9 ng/L (range = 4.5–39.4). We found that sNfL concentrations varied significantly with age (R = 0.48, p < 0.001), but not with BMI (Spearman’s correlation value; −0.18; p = 0.15), EDSS (p = 0.243) or gender (p = 0.979). After adjusting for age, the mean sNfL (all samples) was 11 ng/L (95% CI = 9.6–12.3).
The sNfL variability in EID4–6 was low, as illustrated in Figure 4. The mean individual SD (age adjusted) was 1.55 ng/L (SD = 1.12), and the mean age adjusted individual range was 4.9 ng/L (95% CI = 3.92–5.88).
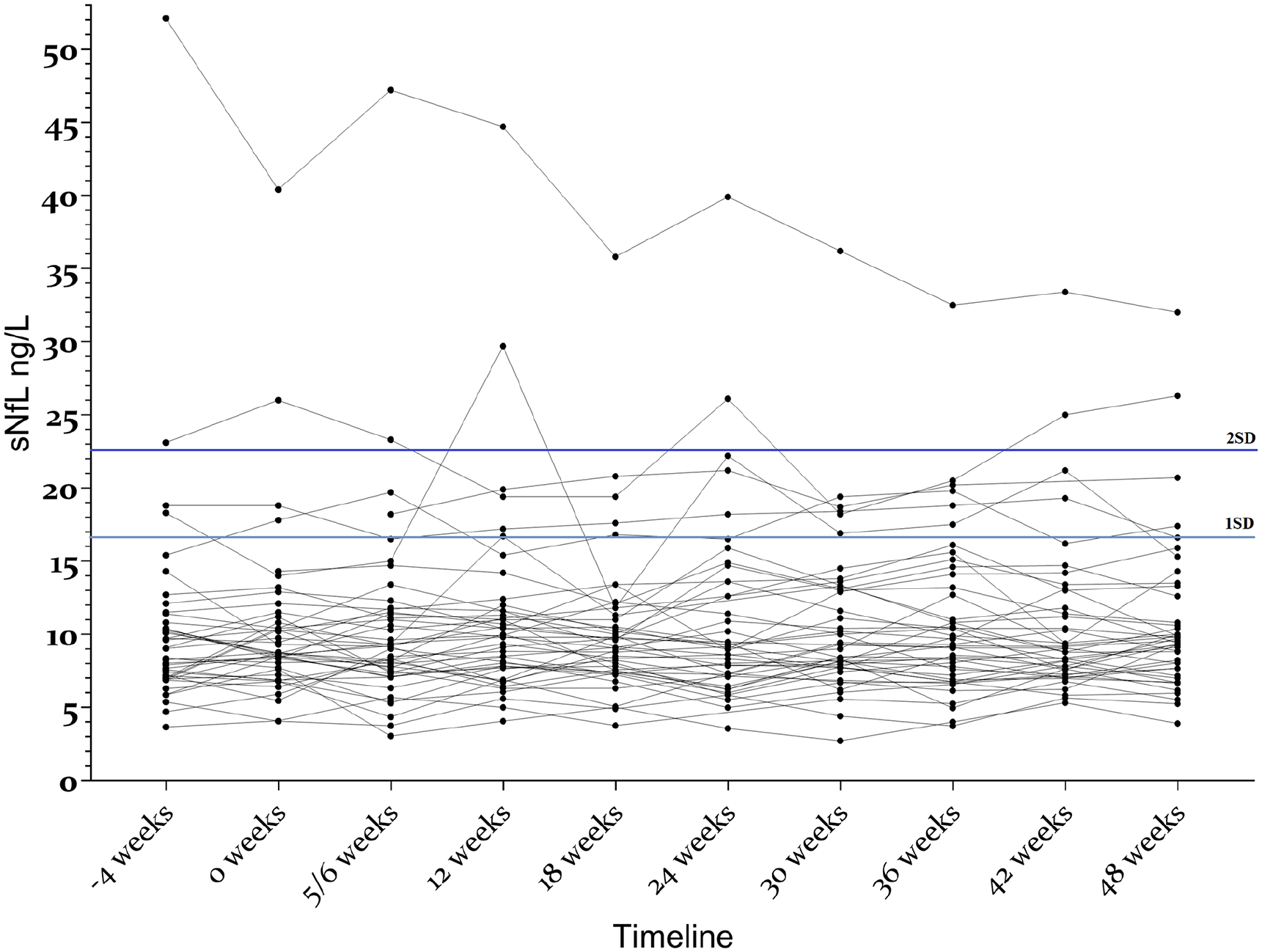
Variations in individual sNfL concentrations over time in patients with relapsing-remitting MS treated with extended-interval natalizumab dosing (EID). All patients were in the EID4–6 group.
We set the mean + 1 SD (16.7 ng/L) and the mean + 2 SD (22.6 ng/L) as cut offs for abnormality. We found one or more samples above this limit in six patients (47 samples, 10.5%) and three patients (17 samples, 3.8%), respectively. Furthermore, in three patients, all samples were above 1 SD, and in one patient, all samples were above 2 SD. None of these patients had clinical or MRI signs of disease activity or disease progression, but all were within the oldest age quartile (49–73 years) of the EID4–6 group. All patients with sNfL concentrations under or equal to 16.7 ng/L (i.e. 1 SD above the mean) were 48 years or younger. In contrast, 6/13 (46%) older patients had one or more samples with sNfL concentrations above 16.7 ng/L (Supplementary Figure S1).
One patient had remarkably high sNfL levels, with a declining trend, but no signs of clinical disease activity on MRIs or evidence of progression in the medical records.
Serum-plasma NfL
Both sNfL and pNfL were collected from a randomly selected subgroup of the EID4–6 cohort (N = 19). The levels of sNfL and pNfL were highly correlated (R = 0.94; p < 0.001; intra-class correlation coefficient = 0.88). Figure 5 shows a pairwise comparison of sNfL and pNfL concentrations in individual samples. On average, the mean sNfL (10.4 ng/L, SD = 3.6) was 14% higher than the mean pNfL (9.13 ng/L, SD = 3.11; p < 0.001).
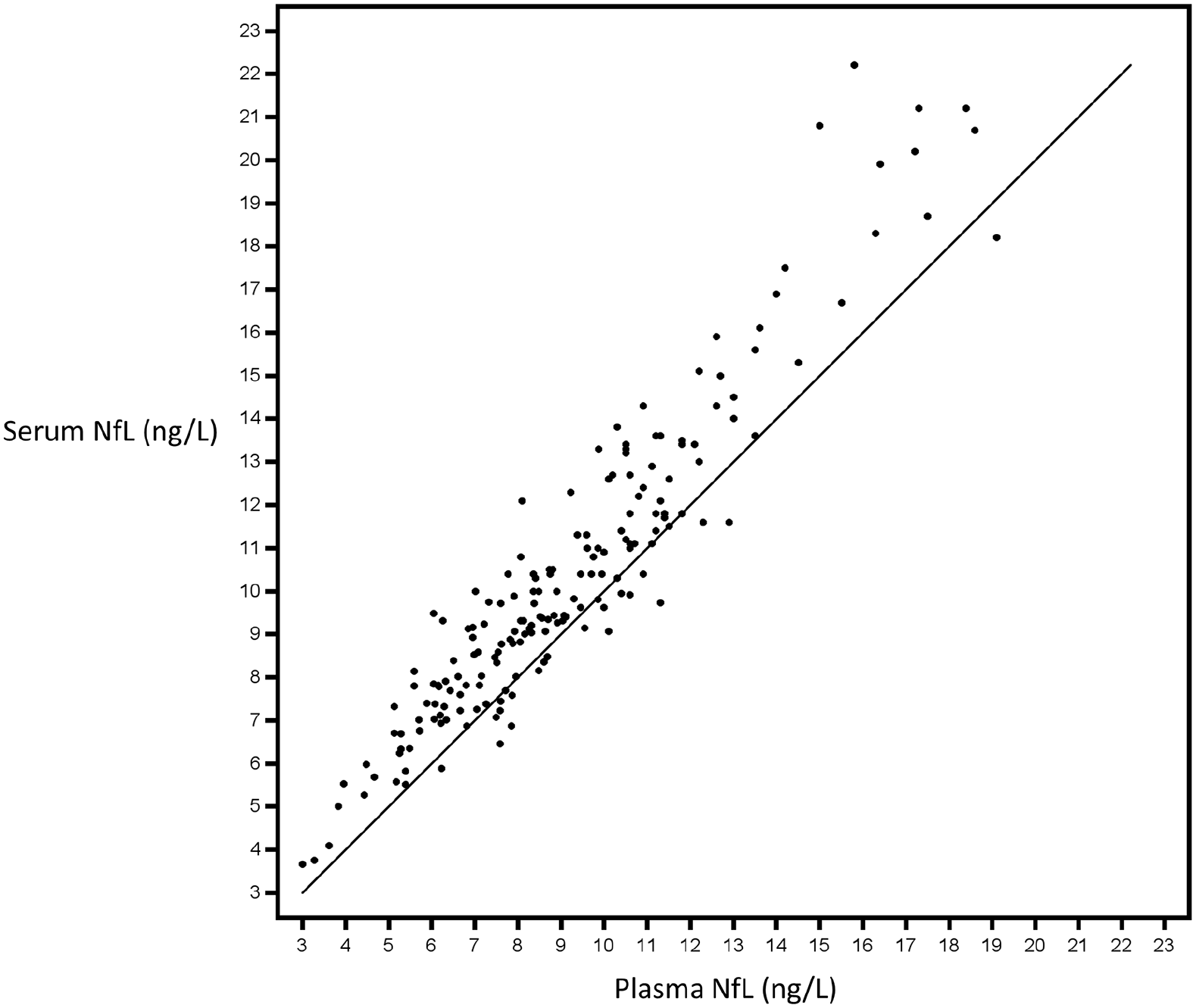
Paired serum and plasma neurofilament light chain (NfL) levels measured in a randomly selected subgroup of patients with relapsing-remitting MS who switched from standard-interval to extended-interval natalizumab dosing (EID). All patients (N = 19) were in the EID4–6 treatment group. Data are from 184 samples. The Shrout-Fleiss reliability random test showed an intra-class correlation coefficient of 0.88; Pearson’s correlation coefficient is R = 0.94 (p = 0.001).
Discussion
The results from this study were consistent with those from previous studies, which showed that extending NZ dosing from 4 to 6 weeks did not affect clinical or MRI measures.6–10 In addition, we showed that the sNfL concentrations were unchanged during 12 months of EID with NZ in patients with RRMS. The NfL concentrations in patients who received SID were similar to those observed in patients who had received EID prior to baseline. These findings supported the notion that axonal damage, determined with sNfL, did not increase when patients were switched to EID with NZ.
The SID of 4 weeks was based on pharmacokinetic and pharmacodynamic properties of NZ. With SID, NZ concentrations are maintained at levels that ensure at least 70%–80% continuous saturation of α4β1 integrin receptors. 30
However, several studies have shown that much lower NZ receptor occupancy was sufficient to block the extravasation of autoreactive immune cells,31,32 the culprit of the CNS attack in RRMS. Previous studies have shown that interruptions longer than 12 weeks in NZ treatment led to increased risk of disease activity.31–35 In contrast, the efficacy provided with 5- to 8-week dosing intervals appeared to be similar to that achieved with SID, when evaluating relapse rate, EDSS, and lesions detected with MRI.6–11,36
However, inflammatory activity and degeneration may escape detection with conventional monitoring.12–14 We have previously shown that NfL and CXCL13 in CSF may be increased in patients without clinical or on MRI signs of disease activity or progression. 14 Moreover, patients with clinically stable RRMS may experience disability deterioration and/or slowly expanding lesions. 13
Even though smoldering MS activity may escape detection by conventional monitoring of RRMS, 12 paramagnetic rim MRI lesions (PRLs) have been associated with chronic or smoldering lesions. 13 Recently, increased sNfL levels were found in RRMS and progressive MS without recent disease activity but with two or more PRLs. 37 Thus, although smoldering MS is considered a slow process, it may give rise to elevations in sNfL levels. In the same study, PRLs were associated with disease severity. In contrast, this study population included only patients with RRMS at baseline and with a few exceptions they did not progress at follow-up. Besides, the observational period was limited to 12 months which probably was too short to detect increases of sNfL due to smoldering MS. However, we cannot rule out that EID with NZ can impact such disease activity.
We monitored sNfL levels to detect new disease activity in patients treated with NZ that switched from SID to EID. Previous studies have used sNfL to evaluate potential disease recurrences in patients who switched from SID to EID. In one previous study, 34 patients with RRMS switched from SID to EID (from 5- to 7-week intervals), and sNfL was essentially unchanged from baseline, after 12 months of NZ treatment. 8 In another study, sNfL remained stable for up to 8 weeks after discontinuing NZ, due to JC virus antibody positivity, but increased sNfL levels were detected at follow-up. 38 In a group of 60 stable MS patients on SID NZ treatment, no change in sNfL was seen at 6 months after switch to ⩾35 days dosing interval. 39 However, our study was the first to monitor sNfL at a relatively high frequency (every 6 weeks) for an extended period of time (48 weeks).
Repeated serum sampling provided the means to investigate treatment effects at both the individual and group levels over time. Due to the temporal change of NfL after a relapse or CNS injury, the interval between testing sNfL should not exceed 3–6 months.20,40 When we determined sNfL concentrations every 6 weeks, we found that most NZ-treated patients had only minor fluctuations of sNfL. We also found that sNfL concentrations were slightly, but significantly higher than pNfL concentrations, consistent with findings from a previous study. 41 With a few exceptions, included patients had essentially no disease activity or disability progression and 94% achieved NEDA-3. Although this selected cohort had apparently very low disease activity prior baseline and during the 48-week study period, approximately 11% and 4% of samples had sNfL concentrations greater than 1 SD and 2 SD, respectively, above the mean sNfL. However, most of those samples were obtained from a few patients who had relatively high sNfL levels during the entire study period. In these patients, the elevated sNfL levels were not associated with disease activity or progression. Among the six patients with sNfL values above 1 SD, two had diabetes, which is known to influence sNfL levels, 42 and all six patients were within the oldest quartile of the EID4–6 group. Similarly, in a previous study, the sNfL variance was higher among individuals of older age, in a population of patients with RRMS who had been followed with repeated serum sampling. 43 The sNfL inter-individual variability seemed larger than the intra-individual variability, suggesting that the best utility of sNfL measurements is for individual longitudinal follow-up of younger adult age. Nevertheless, in our cohorts, some patients displayed unexplained variations in sNfL levels, which suggested the possibility that subtle disease activity could have occurred, but was not detected with conventional monitoring. 12
This study had some limitations. One limitation was the relatively small number of patients included, preventing us from establishing a valid age- and BMI-adjusted reference sNfL concentration in stable RRMS. Although some population-based surveys of healthy subjects have provided age- and BMI-adjusted sNfL concentrations,41,43 there is an unmet need for data on sNfL variability in clinically meaningful subgroups of patients with MS. Perhaps the most important such cohort would be patients with RRMS that receive disease-modifying treatment, particularly those with stable disease monitored with conventional clinical and MRI measures. Our results from repeated sNfL determinations, in most cases, confirmed the stability of clinical and MRI measurements, but individual deviations could occur. A second limitation was that, in contrast to previous studies,19,20,44 we identified new disease activity in only one patient. Thus, we could not investigate the utility of sNfL testing for detecting disease activity in our patient cohort. Another limitation was the lack of healthy control subjects to serve as a reference for comparing sNfL levels to those found in patients with stable disease under NZ treatment. However, the sNfL levels of this study population were similar to those observed in healthy control subjects in a previous study of ours, using similar Simoa assay, 17 but slightly higher than those reported in other studies.41,43
In conclusion, based on repeated determinations of sNfL measurements over 48 weeks, we did not find any signs of increased axonal damage in patients who received NZ and switched from SID to EID. Our data supported the notion that sNfL monitoring was most reliable for monitoring younger adult patients with RRMS, while increased sNfL concentrations may still occur in stable RRMS where confounding factors such as comorbidities are more common.
Supplemental Material
sj-tif-1-msj-10.1177_13524585221108080 – Supplemental material for No increase of serum neurofilament light in relapsing-remitting multiple sclerosis patients switching from standard to extended-interval dosing of natalizumab
Supplemental material, sj-tif-1-msj-10.1177_13524585221108080 for No increase of serum neurofilament light in relapsing-remitting multiple sclerosis patients switching from standard to extended-interval dosing of natalizumab by Magnus Johnsson, Helen H Farman, Kaj Blennow, Henrik Zetterberg, Clas Malmeström, Markus Axelsson and Jan Lycke in Multiple Sclerosis Journal
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.J. has no conflict of interest. H.H.F. has no conflict of interest. H.Z. has served on scientific advisory boards and/or as a consultant for AbbVie, Alector, Annexon, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, NervGen, Novo Nordisk, Pinteon Therapeutics, Red Abbey Labs, Passage Bio, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave. H.Z. has also given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche. H.Z. is also a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is part of the GU Ventures Incubator Program (outside submitted work). K.B. has served as a consultant, on advisory boards, or on data monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD/Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Ono Pharma, Pharmatrophix, Prothena, Roche Diagnostics, and Siemens Healthineers. KB is also a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program, outside the work conducted for this study. C.M. has served on scientific advisory boards and/or held lectures for Biogen, Merck, Novartis, Roche, and Sanofi. C.M. also serves as board member for Alzinova AB, Sweden. M.A. has served on scientific advisory boards and/or held lectures for Biogen, Merck, Novartis, Roche, and Sanofi. J.L. has received lecture honoraria from Biogen, BMS, Merck, Novartis, Sanofi, Roche, and Alexion. J.L. has also served on scientific advisory boards for Biogen, BMS, Merck, Novartis, Sanofi, Roche, and Alexion. J.L. serves on the editorial board of the Acta Neurologica Scandinavica, and has received unconditional research grants from Biogen and Novartis.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the Berit Linnea and Ragnar Bakken Foundation, the Swedish Research Council (#2017-00915); the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615); the Swedish Alzheimer Foundation (#AF-930351, #AF-939721 and #AF-968270); Hjärnfonden, Sweden (#FO2017-0243 and #ALZ2022-0006); the Swedish state under the agreement between the Swedish government and the County Councils; the ALF-agreement (#ALFGBG-715986 and #ALFGBG-965240); the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236); the National Institute of Health (NIH), USA (grant #1R01AG068398-01) and the Alzheimer’s Association 2021 Zenith Award (ZEN-21-848495); the Swedish Research Council (#2018-02532); the European Research Council (#681712); the Swedish State Support for Clinical Research (#ALFGBG-71320); USA (#201809-2016862); the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376C, #ADSF-21-831381C, and #ADSF-21-831377C); the Olav Thon Foundation; the Erling-Persson Family Foundation; Stiftelsen för Gamla Tjänarinnor; Hjärnfonden, Sweden (#FO2019-0228); the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 860197 (MIRIADE); the European Union Joint Programme—Neurodegenerative Disease Research (JPND2021-00694); and the UK Dementia Research Institute at UCL (UKDRI-1003).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.