
Abstract
Background:
Ofatumumab, the first fully human anti-CD20 monoclonal antibody, is approved in several countries for relapsing multiple sclerosis (RMS).
Objective:
To demonstrate the bioequivalence of ofatumumab administered by an autoinjector versus a pre-filled syringe (PFS) and to explore the effect of ofatumumab on B-cell depletion.
Methods:
APLIOS (NCT03560739) is a 12-week, open-label, parallel-group, phase-2 study in patients with RMS receiving subcutaneous ofatumumab 20 mg every 4 weeks (q4w) (from Week 4, after initial doses on Days 1, 7, and 14). Patients were randomized 10:10:1:1 to autoinjector or PFS in the abdomen, or autoinjector or PFS in the thigh, respectively. Bioequivalence was determined by area under the curve (AUC τ ) and maximum plasma concentration (Cmax) for Weeks 8–12. B-cell depletion and safety/tolerability were assessed.
Results:
A total of 256 patients contributed to the bioequivalence analyses (autoinjector-abdomen, n = 128; PFS-abdomen, n = 128). Abdominal ofatumumab pharmacokinetic exposure was bioequivalent for autoinjector and PFS (geometric mean AUC τ , 487.7 vs 474.1 h × µg/mL (ratio 1.03); Cmax, 1.409 vs 1.409 µg/mL (ratio 1.00)). B-cell counts (median cells/µL) depleted rapidly in all groups from 214.0 (baseline) to 2.0 (Day 14). Ofatumumab was well tolerated.
Conclusion:
Ofatumumab 20 mg q4w self-administered subcutaneously via autoinjector is bioequivalent to PFS administration and provides rapid B-cell depletion.
Keywords
Introduction
Disease-modifying therapies (DMTs) for multiple sclerosis (MS) include anti-CD20 monoclonal antibodies (mAbs). These highly effective treatments target B-cells and a small subset of CD20-expressing T-cells, causing their rapid depletion.1–3 Various anti-CD20 mAbs, such as rituximab, ocrelizumab, ublituximab, and ofatumumab, demonstrate differences in epitope binding and induction of immune response.4,5
Ofatumumab is the first fully human anti-CD20 mAb for the treatment of MS and, owing to its unique binding site, has a mode of action that is different to those of other anti-CD20 mAbs. Ofatumumab binds to two distinct, non continuous regions on a unique conformational epitope of the CD20 receptor, including the small and large extracellular loops. 6 This results in a slower off-rate and an increased binding affinity compared with previously developed anti-CD20 mAbs. 1 These properties allow induction of B-cell lysis, primarily occurring through pronounced complement-dependent cytotoxicity and efficient B-cell depletion.6–8 Ofatumumab has similar clinical efficacy to, and more favorable safety and tolerability profiles than other highly efficacious mAb DMTs (i.e. alemtuzumab, natalizumab, and ocrelizumab).2,9–13
Previously approved anti-CD20 mAb treatments cannot be easily and conveniently used by the patient at home and are typically administered in infusion centers for ease of safety monitoring. 14 In contrast, ofatumumab has been developed for subcutaneous self-administration at home at monthly doses of 20 mg (in 0.4 mL). 1
In the phase-3 ASCLEPIOS I/II trials in patients with relapsing multiple sclerosis (RMS), ofatumumab 20 mg administered subcutaneously every 4 weeks (q4w) was superior to oral teriflunomide 14 mg once daily, with greater reductions in relapse rates, close to complete abrogation of focal inflammatory disease activity on magnetic resonance imaging (MRI), and greater delays in disability worsening. 1 The majority of patients self-injected ofatumumab at home with a pre-filled syringe (PFS) after training and following the initial doses, which were administered under medical supervision. 1
Subcutaneous self-administration of ofatumumab in a nonclinical setting may offer advantages for patients, such as flexibility and convenience, as well as time and cost savings, owing to the avoidance of infusion procedures. An autoinjector (AI) pen could help facilitate self-administration at home, thus further reducing treatment burden on patients.
APLIOS was a 12-week, open-label, phase-2 study in patients with RMS designed to test bioequivalence between ofatumumab 20 mg q4w administered subcutaneously by a PFS and by an AI pen. The course of B-cell depletion associated with ofatumumab treatment and MRI lesion activity were explored to characterize the effects of ofatumumab further.
Methods
Trial oversight: standard protocol approvals, registrations, and patient consents
The APLIOS study (NCT03560739) was conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice, 15 local regulatory requirements, and the principles of the Declaration of Helsinki. 16 The study protocol was approved by an independent ethics committee or institutional review board for each study site, and all patients provided written informed consent before commencing trial-related procedures.
Study design and patients
APLIOS was a randomized, open-label, multicenter, parallel-group, 12-week, phase-2 study conducted at 30 centers in nine countries (Austria, Bulgaria, Czech Republic, Estonia, Latvia, Lithuania, Russian Federation, Spain, and the United States). A parallel-group, rather than a traditional crossover pharmacokinetic (PK), study design was used to avoid potential carry-over treatment effects.
Key inclusion criteria were as follows: age of 18–55 years; a diagnosis of MS according to the 2010 revised McDonald criteria, 17 and RMS (relapsing–remitting MS or secondary progressive multiple sclerosis (SPMS) with disease activity) as defined by Lublin et al.; 18 an Expanded Disability Status Scale score of 0–5.5 (inclusive); a history of at least one relapse or a gadolinium-enhancing (Gd+) T1 lesion in the previous year, or of two relapses in the previous 2 years; and neurologically stable disease in the month before randomization.
Key exclusion criteria were as follows: primary progressive MS or SPMS without disease activity; immune system diseases other than MS; an immunodeficiency syndrome; active systemic infections; and suspected or confirmed progressive multifocal leukoencephalopathy. Pregnant or lactating women and women of childbearing potential, unless using highly effective methods of contraception, were also excluded. A full list of exclusion criteria is given in the Supplementary Appendix.
Study procedures
Using interactive response technology, eligible patients were randomly assigned in a 10:10:1:1 ratio, respectively, to one of the following four groups (all using 20 mg subcutaneous injection): (1) AI-abdomen—injection with AI pen administered in the abdomen; (2) PFS-abdomen—injection with PFS administered in the abdomen; (3) AI-thigh—injection with AI pen administered in the thigh; and (4) PFS-thigh—injection with PFS administered in the thigh. The primary comparison was between AI-abdomen and PFS-abdomen. Comparison between abdomen and thigh groups was performed for PK profiles, with no formal testing. Randomization was stratified by body weight (<60, 60–90, and >90 kg) to ensure a balanced distribution between groups, thus limiting the potential effect of weight on the primary endpoints in this parallel-group design.
All patients received open-label ofatumumab for 12 weeks, starting with initial 20 mg doses on Days 1, 7, and 14, followed by subsequent dosing of 20 mg q4w from Week 4 onwards (on Days 28, 56, and 84). Key assessment schedules included PK sampling on Days 1, 4, 7, 14, 28, 42, 56, 57, 59, 63, 70, 77, and 84, with PK samples from Days 56–84 (Weeks 8–12) being used for bioequivalence testing; immunogenicity at baseline and on Days 28, 56, and 84; B-cell counts at baseline and on Days 1, 4, 7, 14, 28, 42, 56, and 84; and MRI scans at screening and on Days 28, 56, and 84.
At the end of Week 12 (study completion), eligible patients could continue treatment with ofatumumab by enrolling in an “umbrella,” open-label, phase-3b, extension study (ALITHIOS (NCT03650114)). Patients who did not enter the extension study (n = 3) were followed up every 3 months for at least 9 months or until B-cell repletion (i.e. until levels returned to baseline values or to the lower limit of normal (LLN, 80 cells/µL), whichever came first) after study-drug discontinuation. Further information on B-cell repletion following cessation of ofatumumab treatment will be collected as part of the ongoing ALITHIOS study.
Study objectives and outcome measures
The primary objective was to demonstrate bioequivalence for injections of ofatumumab 20 mg in the abdomen administered via AI pen and those administered via PFS. The primary endpoints were area under the plasma concentration–time curve over the dosing interval (AUC τ ) and maximum plasma concentration (Cmax) following drug administration. Assessment of PK endpoints was based on data collected between Weeks 8 and 12, during which approximate steady-state PK was anticipated. Plasma concentrations of ofatumumab were determined by validated enzyme-linked immunosorbent assay. A modified reference-scaled average bioequivalence approach was used to establish bioequivalence, as described in the statistical analysis section.
Secondary objectives included (1) comparison of PK profiles for ofatumumab injection in the abdomen versus the thigh; (2) assessment of ofatumumab immunogenicity; and (3) assessment of the safety and tolerability of ofatumumab. Endpoints for the secondary objectives included: (1) AUC τ and Cmax; (2) proportion of patients with anti-ofatumumab antibodies (analyzed by a Meso Scale Discovery electrochemiluminescence assay); and (3) adverse events (AEs), including injection-related systemic reactions (i.e. systemic reactions occurring in the 24 hours after an injection) and injection site reactions (i.e. reactions localized to the injection site), abnormalities in vital signs, laboratory evaluations and electrocardiograms, and electronic Columbia Suicide Severity Rating Scale scores. AE severity was graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 5 (except change in serum amylase, which was assessed using version 4.03). 19
Exploratory endpoints included depletion of CD19+ B cells (as a surrogate for CD20+ B cells) and MRI parameters (including new or persistent Gd+ T1 lesions).
Statistical analyses
A sample size of 150 patients (124 in the abdomen groups and 26 in the thigh groups) was calculated to provide at least 90% power for bioequivalence testing between the PFS-abdomen and AI-abdomen groups, assuming a PK variability not exceeding 85%, as measured by the coefficient of variation (CV). An interim analysis was planned to assess PK variability and to provide an opportunity to increase the sample size, if needed, so that sufficient power for bioequivalence testing could be achieved even with variability being higher than anticipated. After review of the CV for the initial 36 patients, the total sample size was increased to 284 patients, randomly assigned to the AI-abdomen (n = 128), PFS-abdomen (n = 130), AI-thigh (n = 13), and PFS-thigh (n = 13) groups. This was estimated to provide approximately 80% power for bioequivalence testing for CVs up to approximately 240%.
Analysis of bioequivalence between abdominal PFS and AI pen administration was conducted using the reference-scaled average bioequivalence approach for highly variable drugs, which is recommended by the US Food and Drug Administration,20,21 modified for a parallel-group study design. Bioequivalence was declared if the approximate 95% upper confidence bound of the linearized criterion was equal to, or less than, 0 and if the geometric mean ratio was contained within the predefined limits (0.80–1.25). As per the protocol, bioequivalence testing was performed for AUC τ and Cmax separately on all patients randomized to the PFS-abdomen and AI-abdomen groups with valid PK data during the dosing interval (Weeks 8–12; n = 256).
For the comparison of PK endpoints between abdominal and thigh injection locations, sample size requirements (26 patients, 13 per group) were based on the conventional size of studies of this type and considered dropout rate (i.e. no formal statistical testing was planned).
For secondary endpoints, summary statistics of ofatumumab plasma concentrations by time point were provided for the four groups contributing to the PK analysis (i.e. all randomized patients with PK data during dose administration). Safety analyses, as well as analyses of B-cell and Gd+ T1 lesion data, were also conducted in all randomized patients who received at least one dose of study drug.
Statistical methods used in this study will be described in detail in a separate article.
Data availability statement
The APLIOS data are available on reasonable request, provided that the request is in line with current ethical and intellectual property requirements surrounding the use of data. Requests should be directed through ClinicalStudyDataRequest.com.
Results
Of the 344 patients screened, 284 were randomly assigned to open-label ofatumumab in one of the four groups. Most screening failures (57/60) were due to patients not meeting inclusion/exclusion criteria. In total, 258 patients (i.e. both abdomen groups) were initially planned for inclusion in the bioequivalence analysis but, given that two patients missed the Week-8 dose, data from 256 patients (PFS-abdomen: n = 128; AI-abdomen: n = 128) were included in the bioequivalence testing. Data from all 284 randomized patients were included in the PK, safety, B-cell, and MRI analyses. Nearly all patients (n/N = 283/284, >99%) completed the study; one patient in the PFS-abdomen group discontinued owing to an AE (Figure 1). Baseline demographics and disease characteristics were broadly similar between the PFS-abdomen and AI-abdomen groups (Table 1).
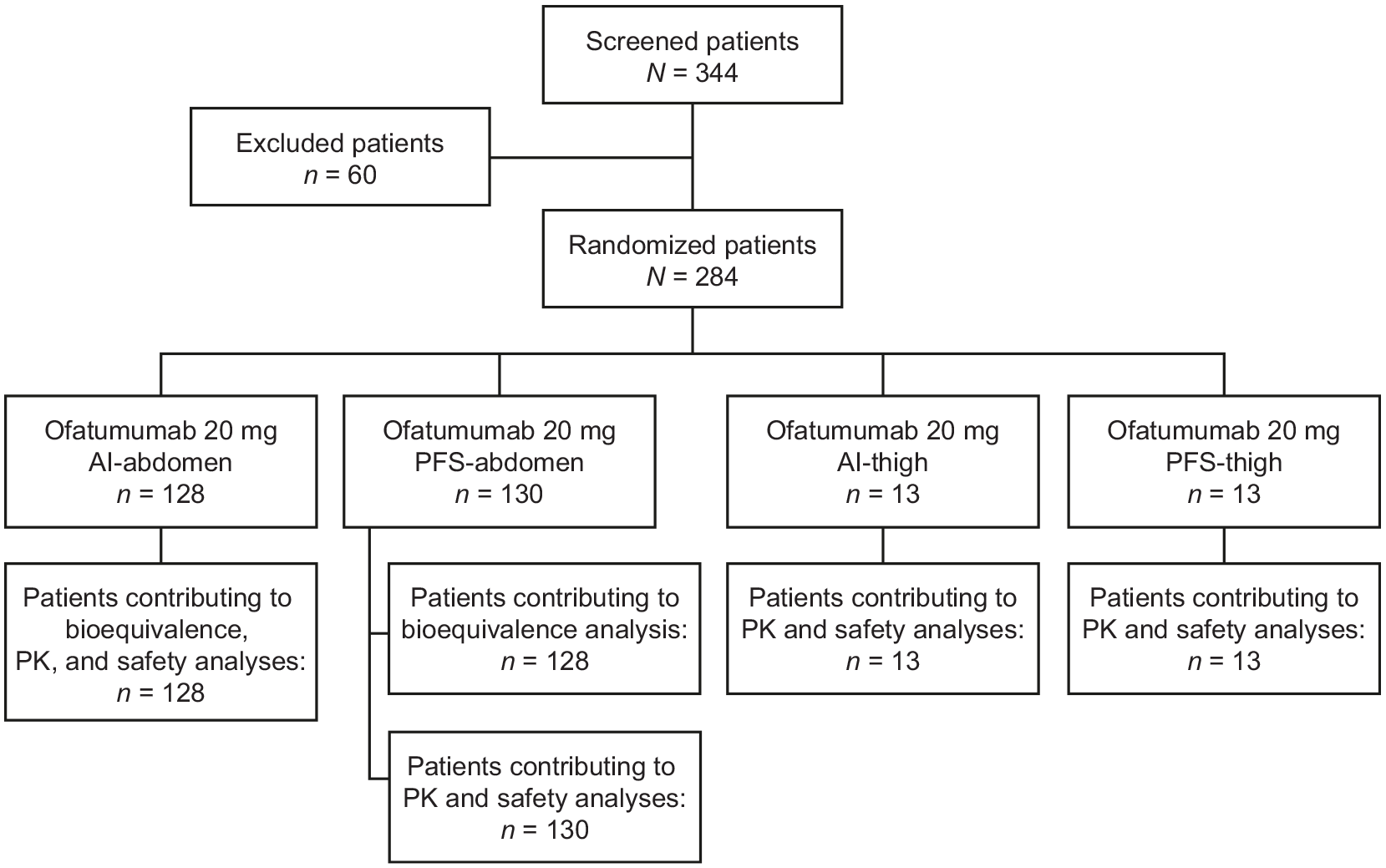
APLIOS patient flow chart. One patient in the PFS-abdomen group discontinued the study following a Grade-2 AE (blood IgM level decreased). Two patients in the PFS-abdomen group, including the patient who discontinued the study, did not contribute to the bioequivalence analysis because they missed the dose at Week 8 and had no data available for the dosing interval (Weeks 8–12).
Baseline demographics and disease characteristics.
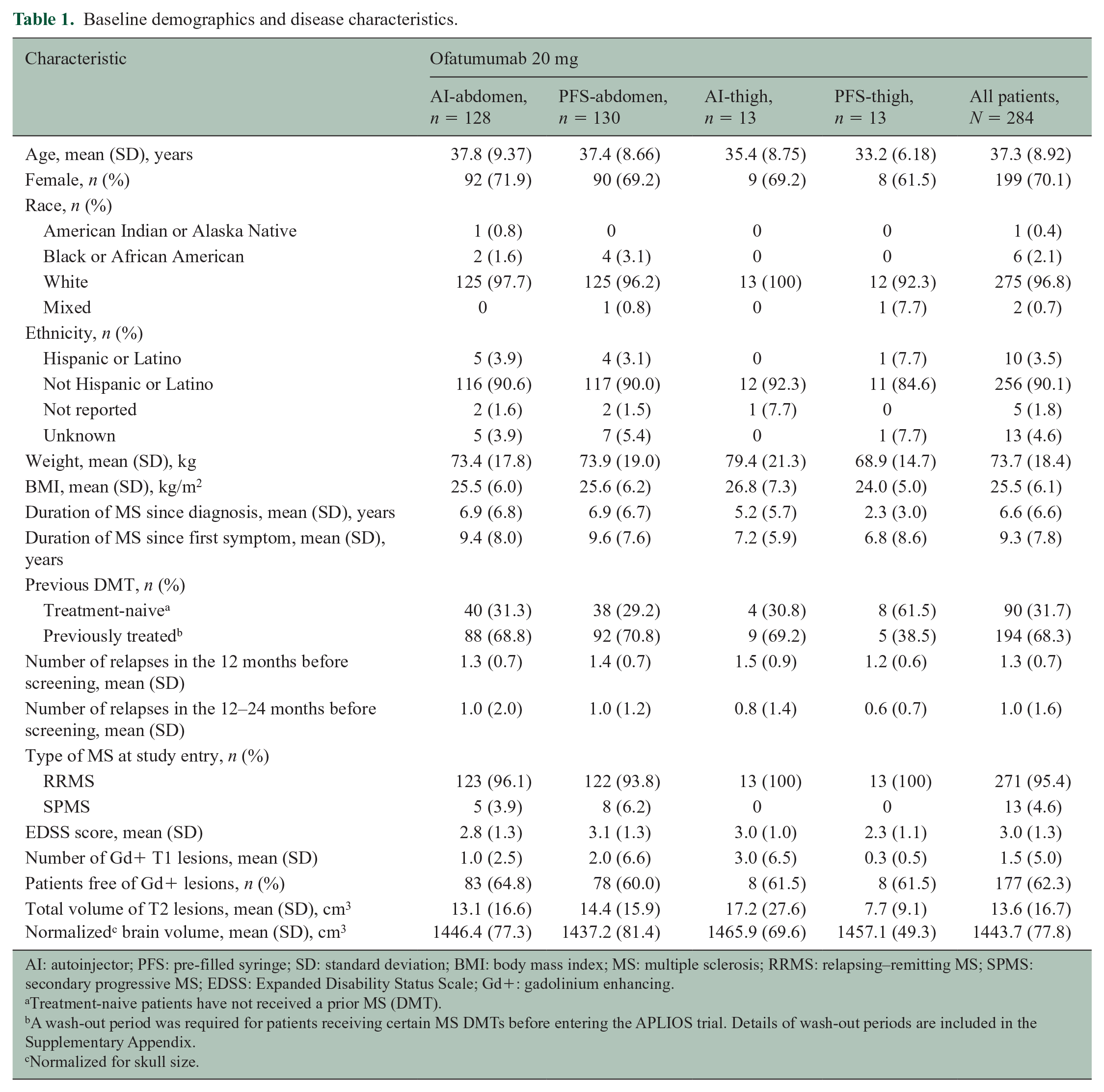
AI: autoinjector; PFS: pre-filled syringe; SD: standard deviation; BMI: body mass index; MS: multiple sclerosis; RRMS: relapsing–remitting MS; SPMS: secondary progressive MS; EDSS: Expanded Disability Status Scale; Gd+: gadolinium enhancing.
Treatment-naive patients have not received a prior MS (DMT).
A wash-out period was required for patients receiving certain MS DMTs before entering the APLIOS trial. Details of wash-out periods are included in the Supplementary Appendix.
Normalized for skull size.
Pharmacokinetics
Primary endpoint: bioequivalence between AI pen and PFS administration
Based on the reference-scaled average bioequivalence approach, abdominal administration of ofatumumab via AI pen showed bioequivalence to that via PFS for both AUC τ and Cmax in the dosing interval (Weeks 8–12; Table 2). The 95% upper bound of the linearized criterion was below 0 for both AUCτ (–0.3131) and Cmax (–0.2446). Geometric mean ratios were contained within the predefined limits (0.8–1.25) for both AUCτ (1.03) and Cmax (1.00).
Bioequivalence testing of AUC τ and Cmax between AI-abdomen and PFS-abdomen during the dosing interval (Weeks 8–12).
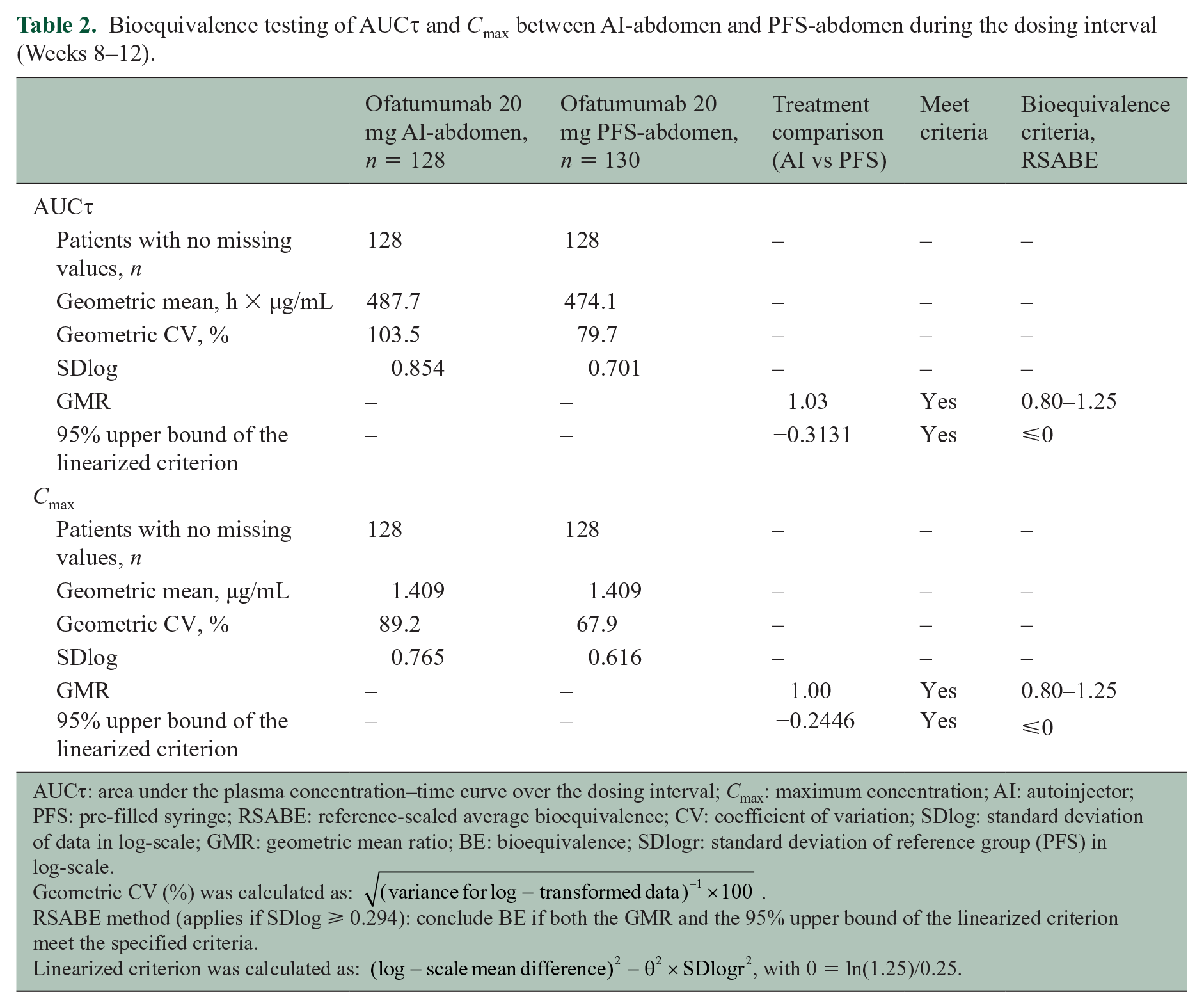
AUCτ: area under the plasma concentration–time curve over the dosing interval; Cmax: maximum concentration; AI: autoinjector; PFS: pre-filled syringe; RSABE: reference-scaled average bioequivalence; CV: coefficient of variation; SDlog: standard deviation of data in log-scale; GMR: geometric mean ratio; BE: bioequivalence; SDlogr: standard deviation of reference group (PFS) in log-scale.
Geometric CV (%) was calculated as:
RSABE method (applies if SDlog ⩾ 0.294): conclude BE if both the GMR and the 95% upper bound of the linearized criterion meet the specified criteria.
Linearized criterion was calculated as:
Comparison of PK by injection site
Plasma concentrations of ofatumumab at each time point were similar across groups regardless of administration device or injection site (Figure 2). Mean AUC τ and Cmax at Week 8 were also similar across groups regardless of administration device or injection site (Table 3).
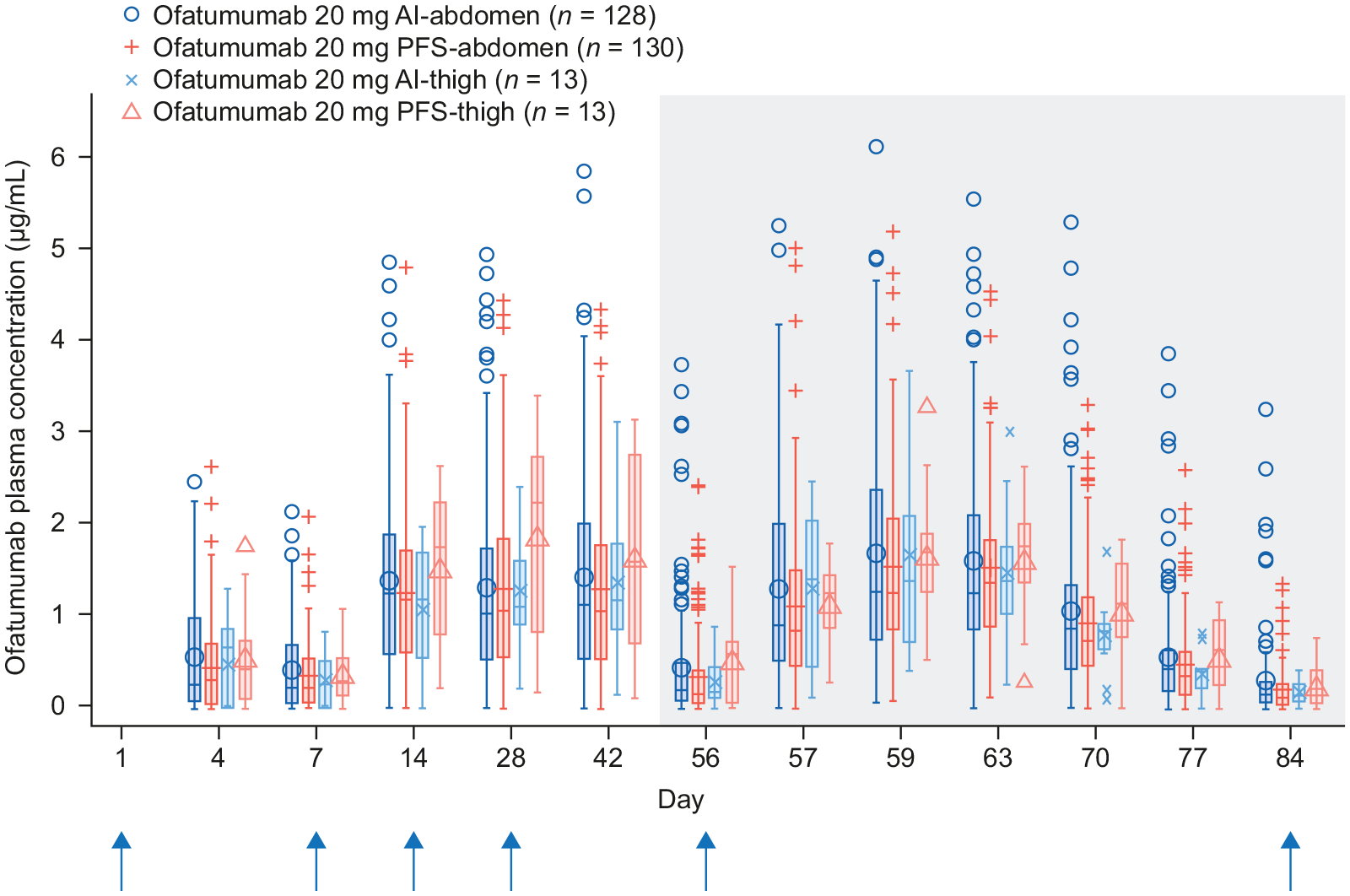
Plasma concentrations of ofatumumab by nominal visit. The blue vertical arrows indicate the timing of dose administration. The shaded region indicates the Week 8−12 dosing interval that was considered for bioequivalence testing.
Summary statistics of AUC τ and Cmax at Week 8 dosing interval.
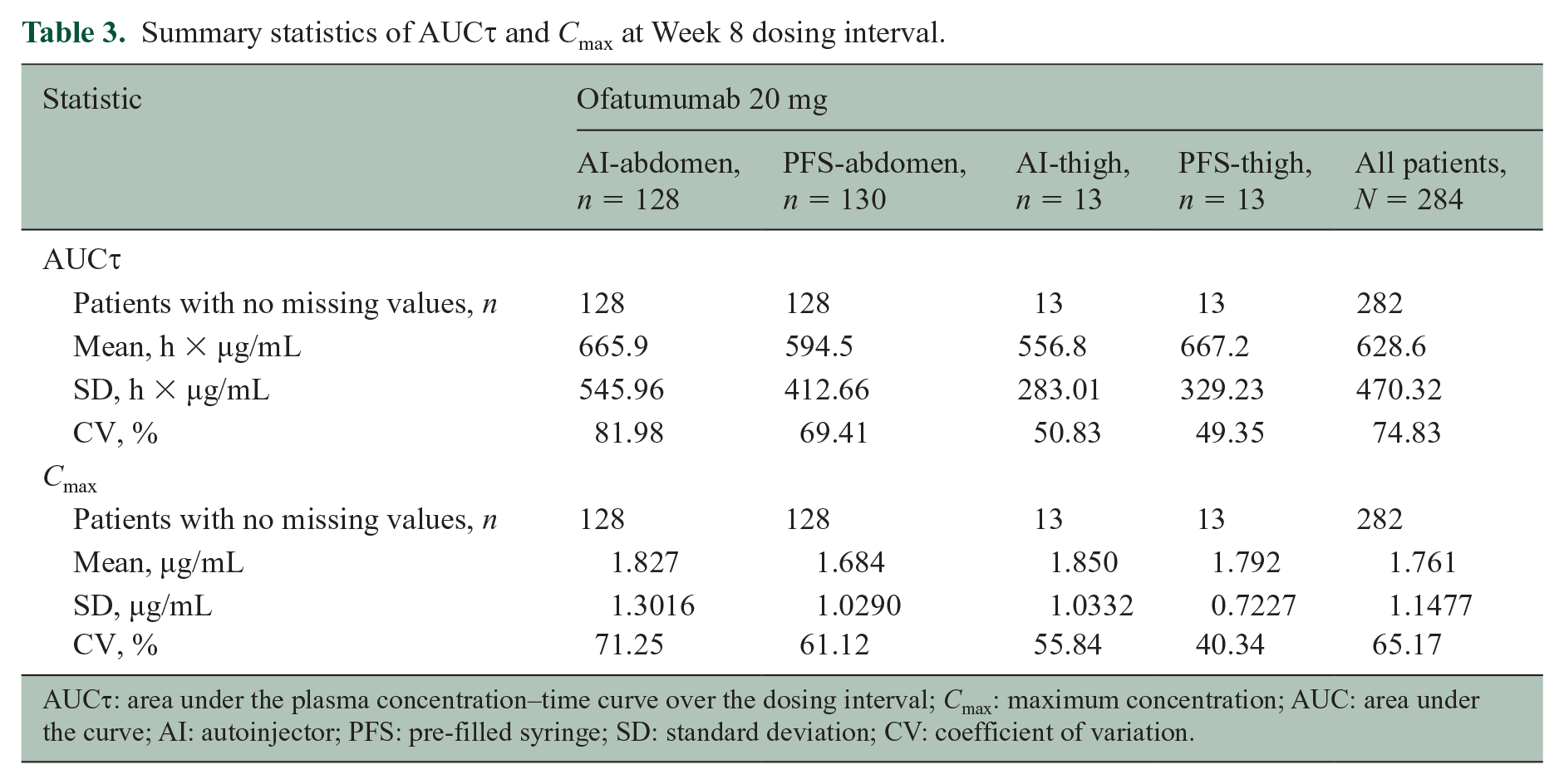
AUC τ : area under the plasma concentration–time curve over the dosing interval; Cmax: maximum concentration; AUC: area under the curve; AI: autoinjector; PFS: pre-filled syringe; SD: standard deviation; CV: coefficient of variation.
Immunogenicity of ofatumumab
Results of ofatumumab anti-drug antibody (ADA) testing were positive in 7/284 patients (2.5%); however, six of these patients had a positive ADA test result at baseline, likely reflecting false-positive findings. Only one patient (0.4%) who did not have a positive ADA test result at baseline had a transiently positive ADA test result at Week 8. There was no observed impact of ADA on PK exposure or B-cell depletion. No patients developed neutralizing antibodies to ofatumumab, as assessed by a cell-based assay.
Safety and tolerability profile
The overall incidence of AEs and serious AEs in the study population was 57.4% and 2.1%, respectively (Table 4). The incidence of AEs leading to study-drug discontinuation was 0.4%. No deaths occurred during the study. Commonly reported AEs (i.e. in ⩾2% of patients) are summarized in Table 4. Injection-related reactions were predominantly observed with the first injection in all groups (Supplemental Figure S1). Injection-related systemic reactions were reported in 25.0% of patients with the first injection, 8.1% of patients with the second injection, and fewer than 2.8% of patients with subsequent injections. The most frequently reported symptoms associated with injection-related systemic reactions were headache, chills, and fever, reported in 12.7%, 8.8%, and 8.5% of patients, respectively. Typically, symptoms resolved within 48 hours for most patients. Injection site reactions were reported in 6.0% of patients with the first injection and ⩽3.2% of patients with subsequent injections. The most frequently reported symptoms associated with injection site reactions were erythema/redness and pain, reported in 5.3% and 3.2% of patients, respectively. Most AEs (97.5%) were CTCAE Grade-1/2 (mild/moderate). The overall incidence of Grade-3 (severe) AEs was low (2.5%); all Grade-3 AEs were isolated events, each occurring in a single patient across the groups. No Grade-4 AEs were reported.
Summary of AEs.
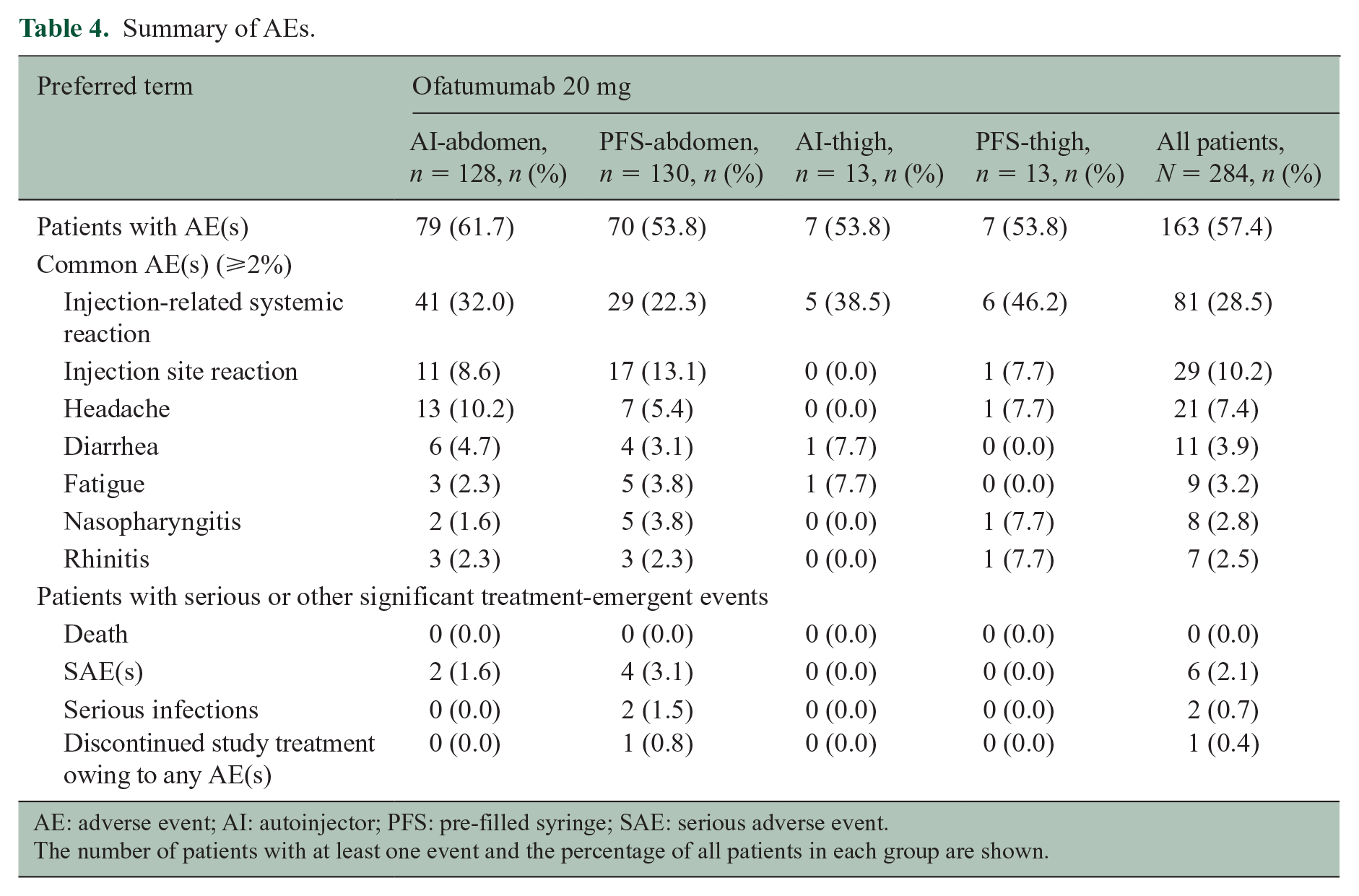
AE: adverse event; AI: autoinjector; PFS: pre-filled syringe; SAE: serious adverse event.
The number of patients with at least one event and the percentage of all patients in each group are shown.
Nearly all injection-related reactions were Grade-1/2 (mild/moderate); there was one Grade-3 (severe) reaction with the first injection in one patient in the PFS-abdomen group. The patient had abdominal pain, arthralgia, asthenia, chills, dizziness, fatigue, fever, headache, myalgia, nausea, and rash (alongside milder site reactions), which lasted for 6 days. The patient continued study treatment per protocol until Week 12 (end of study).
Infections were reported in 58/284 patients (20.4%) overall, with similar incidences across groups. The most commonly reported infections (occurring in ⩾2% of patients overall) were nasopharyngitis (2.8%) and rhinitis (2.5%). Two serious infections (appendicitis and pneumonia, both in the PFS-abdomen group) were reported in one patient each but neither led to study-drug discontinuation. No opportunistic infections were reported.
B-cell depletion
By Day 14, B-cell counts were below the LLN (80 cells/µL) for all patients with available data (n = 273) who were receiving ofatumumab. Median B-cell counts were reduced from 214.0 cells/µL at baseline to 2.0 cells/µL at Day 14 (99.1% depletion) before administration of the third dose. At Week 4 (i.e. between the third and fourth doses), median B-cell count was reduced to 1.0 cells/µL and remained at this level until Week 12 (end of study) (Figure 3(a)). The overall proportion of patients with a B-cell count below 10 cells/µL was 84.6% at Day 14, 94.0% at Week 4, 95.9% at Week 8, and 98.2% at Week 12 (Figure 3(b)). B-cell depletion was similar across all body-weight quartiles (Figure 3(c)).
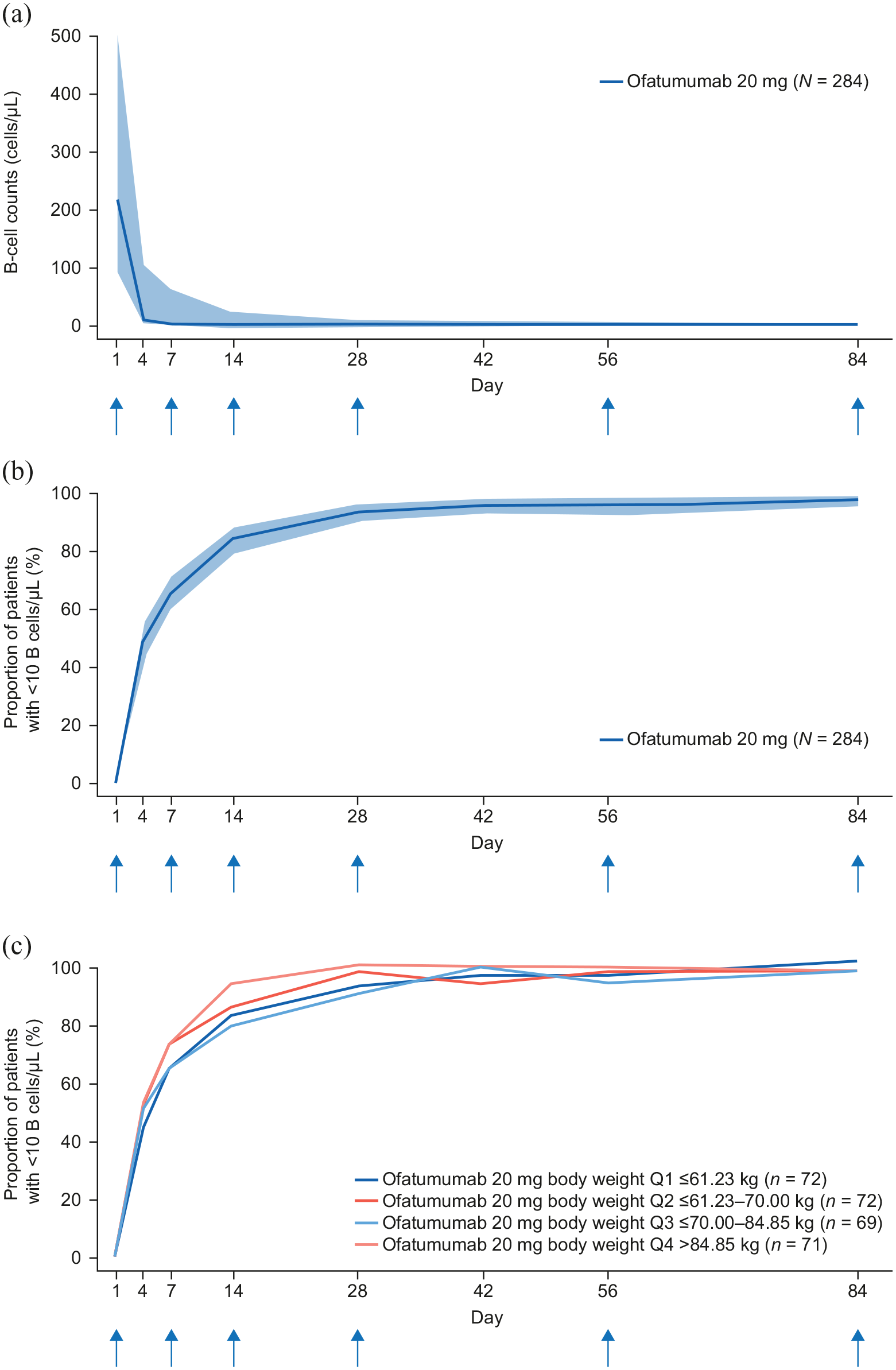
(a) Median number of B-cells over 12 weeks with subcutaneous ofatumumab 20 mg (N = 284), (b) proportion of patients with B-cells < 10 µL over time (N = 284), and (c) proportion of patients with B-cells < 10 µL over time stratified by body-weight quartile. The blue vertical arrows indicate the timing of dose administration. The analysis considered data until 30 days after the last injection. The shaded bands in parts (a) and (b) are 95% confidence intervals calculated using the Clopper–Pearson method.
MRI endpoints
The mean number of new or persisting Gd+ T1 lesions decreased from 1.5 at baseline to 0.8, 0.3, and 0.1 by Weeks 4, 8, and 12, respectively (Figure 4(a)). The proportion of patients free of Gd+ T1 lesions increased over time in all ofatumumab groups (Table 5). Across all groups, the proportions of patients free of Gd+ T1 lesions at baseline and Weeks 4, 8, and 12 were 64.2%, 66.5%, 86.7%, and 94.1%, respectively (Figure 4(b); Table 5).
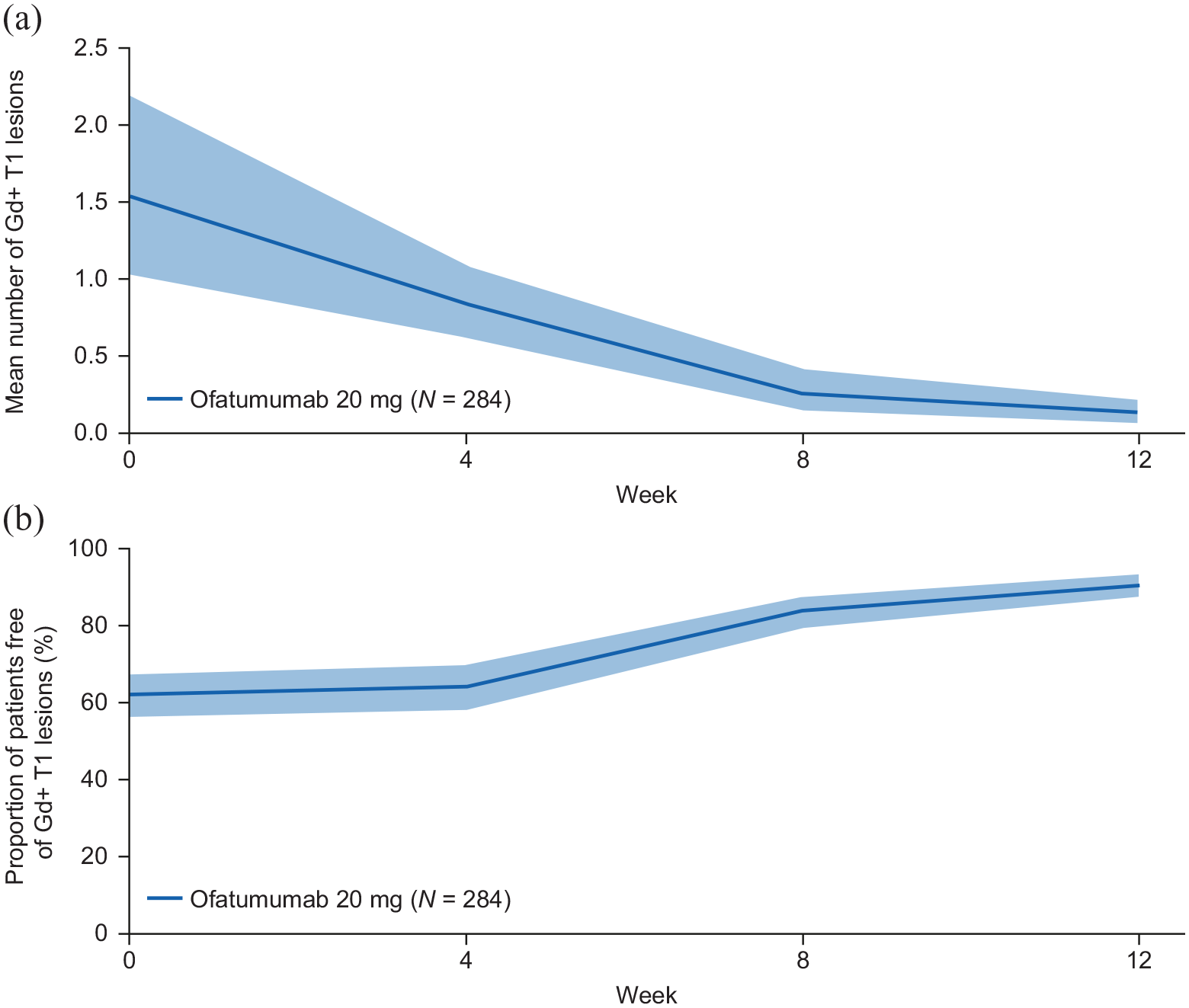
(a) Number of new or persistent Gd+ T1 lesions and (b) proportion of patients free of Gd+ T1 lesions over 12 weeks with ofatumumab treatment. Pre-dose MRI assessments are displayed as time 0 on the x-axis. The analysis considers scans collected until 30 days after the last injection date. Gd+ T1 lesion counts from scans collected in the 14 days after termination of steroid therapy are excluded from the analysis. The shaded bands are 95% confidence intervals calculated (a) from bootstrap and (b) using the Clopper–Pearson method.
Proportion of patients free of Gd+ T1 lesions at Weeks 4, 8, and 12.

Gd+: gadolinium-enhancing; AI: autoinjector; n/M, number of patients who are in the corresponding category/total number of patients with a value for a specific visit window; PFS: pre-filled syringe.
Gd+ T1 lesion counts from scans collected in the 14 days after termination of steroid therapy were excluded from the analysis. The analysis considers scans collected until 30 days after the last injection date.
Discussion
This randomized, open-label, phase-2 APLIOS study demonstrates bioequivalence between ofatumumab 20 mg q4w administered subcutaneously in the abdomen by AI pen and by PFS. Initial dosing with ofatumumab 20 mg on Days 1, 7, and 14 provided rapid B-cell depletion, with subsequent doses q4w maintaining a continuous and almost complete B-cell depletion throughout the study. This rapid and continuous B-cell depletion was seen for all patients regardless of administration device, injection site, or body weight. Notably, the efficient B-cell depletion was accompanied by marked reductions in Gd+ T1 lesion activity as early as Week 8, reaching more than a 90% reduction compared with baseline by Week 12. Alongside the pivotal phase-3 ASCLEPIOS I/II studies, APLIOS provides further evidence for the rapid abrogation of new inflammatory disease activity with ofatumumab. Systemic ofatumumab exposure was similar between injection sites (abdomen or thigh). The number of injection site reactions following administration via AI pen was slightly lower than that following administration via PFS, which may influence patient satisfaction (although this was not assessed in the APLIOS study).
The sustained B-cell suppression with ofatumumab, regardless of body weight, was consistent with that observed in the ASCLEPIOS I/II studies. 1 Moreover, the sustained B-cell depletion associated with ofatumumab was achieved at low systemic drug exposure levels, which remained relatively stable between doses. In contrast, a high-dose intravenous anti-CD20 mAb administered every 6 months showed greater overall fluctuations in plasma drug levels, with initially high concentrations decreasing over time, thus potentially allowing partial B-cell repletion between doses. 22 In animal models, subcutaneous administration of antibodies, compared with infusion, has been shown to permit more direct access to the lymph nodes via absorption into the lymphatic system, rather than via the bloodstream,23,24 thus targeting B cells in the lymph nodes directly and sparing those in the spleen. This has been hypothesized to help to preserve immunosurveillance.
In the APLIOS study, ofatumumab had favorable safety and tolerability profiles, in line with those reported in the ASCLEPIOS I/II trials. 1 The majority of injection-related systemic reactions in APLIOS were mild or moderate in severity and mostly associated with the first injection. Injection-related reactions with ofatumumab were less frequent in an indirect comparison with other anti-CD20 treatments that are administered at higher intravenous doses and require premedication, such as ocrelizumab (particularly when comparing early doses). 25 Indeed, in the ASCLEPIOS I/II studies, there were no notable differences between ofatumumab and placebo injections (used in the teriflunomide arm to maintain treatment blinding) in the frequency of injection-related reactions after the first dose. 1 The generally favorable safety profile of ofatumumab may be related to its fully human nature and unique binding site, as well as to the relatively low dose.
Compared with intravenous administration, subcutaneous administration offers several advantages for patients, including that it affords the flexibility and convenience of self-administration at home or elsewhere, without the need to book an infusion appointment or to travel to a clinic. This may be of particular relevance in the context of the current coronavirus disease 2019 (COVID-19) pandemic and any similar future situations. Moreover, subcutaneous administration is likely to be more cost-effective than intravenous administration owing to the resource efficiency of home-based administration. 26 Patient preference for route of drug administration may also have implications for treatment persistence and quality of life. 26 Subcutaneous administration with AI pen, rather than with PFS, is likely to offer further advantages, such as reductions in perceived pain. 27
Conclusion
The APLIOS study demonstrated bioequivalence between ofatumumab administered into the abdomen by AI pen and by PFS, and corroborated previous findings that ofatumumab, given at low monthly doses of 20 mg (after initial dosing on Days 1, 7, and 14), exhibits favorable properties with regard to fast and continuous B-cell depletion in all patients with RMS. The APLIOS study demonstrated that this highly effective MS therapy with favorable safety and tolerability profiles can be administered using an AI device, allowing for home-based self-administration, thus reducing patient burden.
Supplemental Material
sj-docx-1-msj-10.1177_13524585211044479 – Supplemental material for Rapid and sustained B-cell depletion with subcutaneous ofatumumab in relapsing multiple sclerosis: APLIOS, a randomized phase-2 study
Supplemental material, sj-docx-1-msj-10.1177_13524585211044479 for Rapid and sustained B-cell depletion with subcutaneous ofatumumab in relapsing multiple sclerosis: APLIOS, a randomized phase-2 study by Amit Bar-Or, Heinz Wiendl, Xavier Montalban, Enrique Alvarez, Maria Davydovskaya, Silvia R Delgado, Evgeniy P Evdoshenko, Natasa Giedraitiene, Katrin Gross-Paju, Sulev Haldre, Craig E Herrman, Guillermo Izquierdo, Guntis Karelis, Fritz Leutmezer, Miroslav Mares, Jose E Meca-Lallana, Dalia Mickeviciene, Jacqueline Nicholas, Derrick S Robertson, Denis V Sazonov, Kenneth Sharlin, Bharathy Sundaram, Natalia Totolyan, Marta Vachova, Martin Valis, Morten Bagger, Dieter A Häring, Inga Ludwig, Roman Willi, Martin Zalesak, Wendy Su, Martin Merschhemke and Edward J Fox in Multiple Sclerosis Journal
Footnotes
Appendix
Author contributions.
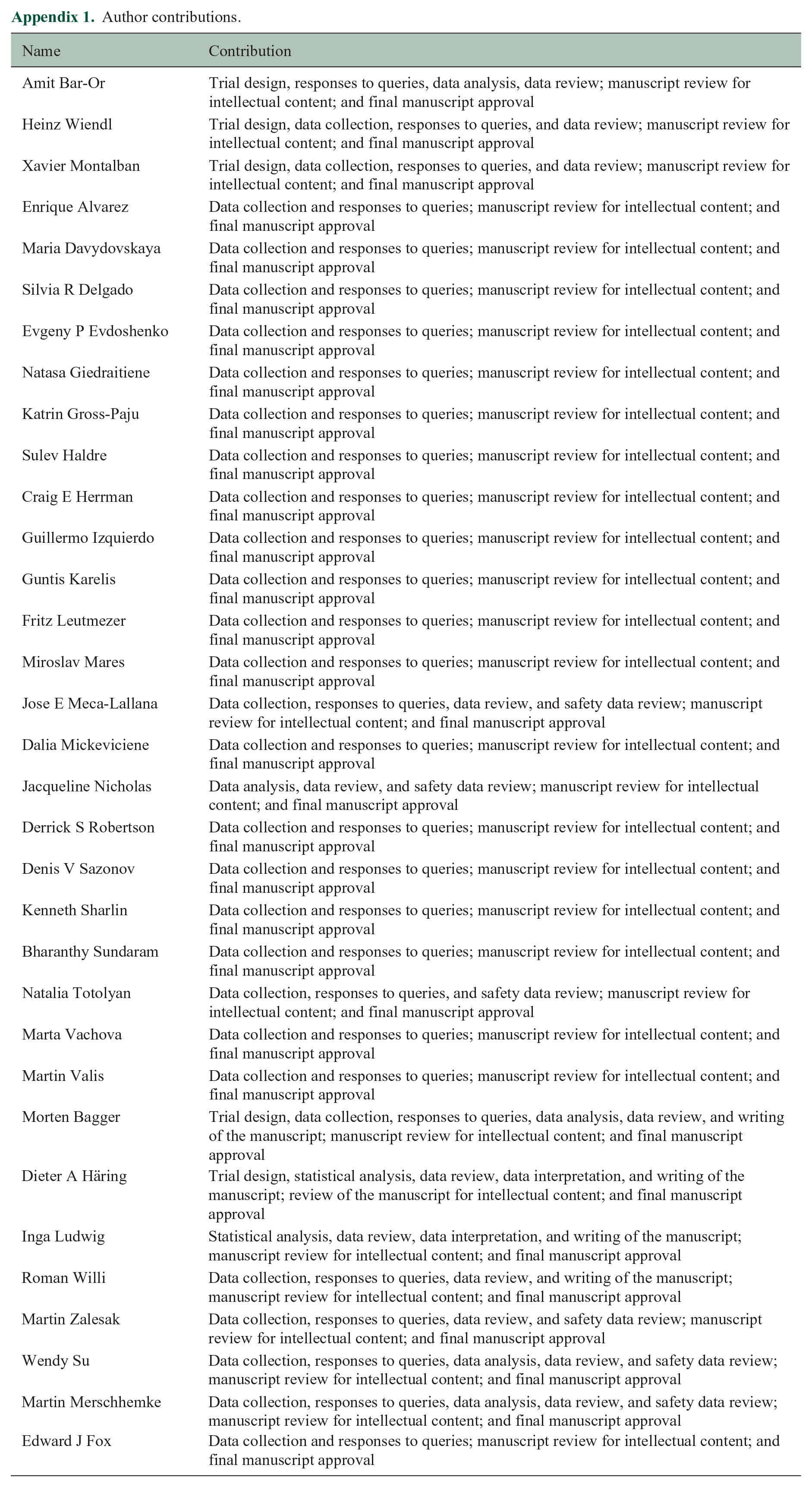
| Name | Contribution |
|---|---|
| Amit Bar-Or | Trial design, responses to queries, data analysis, data review; manuscript review for intellectual content; and final manuscript approval |
| Heinz Wiendl | Trial design, data collection, responses to queries, and data review; manuscript review for intellectual content; and final manuscript approval |
| Xavier Montalban | Trial design, data collection, responses to queries, and data review; manuscript review for intellectual content; and final manuscript approval |
| Enrique Alvarez | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Maria Davydovskaya | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Silvia R Delgado | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Evgeny P Evdoshenko | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Natasa Giedraitiene | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Katrin Gross-Paju | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Sulev Haldre | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Craig E Herrman | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Guillermo Izquierdo | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Guntis Karelis | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Fritz Leutmezer | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Miroslav Mares | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Jose E Meca-Lallana | Data collection, responses to queries, data review, and safety data review; manuscript review for intellectual content; and final manuscript approval |
| Dalia Mickeviciene | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Jacqueline Nicholas | Data analysis, data review, and safety data review; manuscript review for intellectual content; and final manuscript approval |
| Derrick S Robertson | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Denis V Sazonov | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Kenneth Sharlin | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Bharanthy Sundaram | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Natalia Totolyan | Data collection, responses to queries, and safety data review; manuscript review for intellectual content; and final manuscript approval |
| Marta Vachova | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Martin Valis | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
| Morten Bagger | Trial design, data collection, responses to queries, data analysis, data review, and writing of the manuscript; manuscript review for intellectual content; and final manuscript approval |
| Dieter A Häring | Trial design, statistical analysis, data review, data interpretation, and writing of the manuscript; review of the manuscript for intellectual content; and final manuscript approval |
| Inga Ludwig | Statistical analysis, data review, data interpretation, and writing of the manuscript; manuscript review for intellectual content; and final manuscript approval |
| Roman Willi | Data collection, responses to queries, data review, and writing of the manuscript; manuscript review for intellectual content; and final manuscript approval |
| Martin Zalesak | Data collection, responses to queries, data review, and safety data review; manuscript review for intellectual content; and final manuscript approval |
| Wendy Su | Data collection, responses to queries, data analysis, data review, and safety data review; manuscript review for intellectual content; and final manuscript approval |
| Martin Merschhemke | Data collection, responses to queries, data analysis, data review, and safety data review; manuscript review for intellectual content; and final manuscript approval |
| Edward J Fox | Data collection and responses to queries; manuscript review for intellectual content; and final manuscript approval |
Acknowledgements
The authors thank patients for their participation in, and commitment to, APLIOS and the clinical study team for the conduct of the study. The authors are grateful to Xixi Hu and Mikhail Mikhaylov for generating data outputs for this article, to Angela Pozo Ramajo and Karol Bociek (Oxford PharmaGenesis Ltd, Oxford, UK) for providing medical writing support, and to Alessia Gonzato (Oxford PharmaGenesis Ltd, Oxford, UK) for providing editorial support. These services were sponsored by Novartis Pharma AG.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.B-O. reports grants and personal fees from Biogen Idec and Genentech; and personal fees from Atara Biotherapeutics, Brainstorm, Celgene/Receptos, GlaxoSmithKline, Janssen/Actelion, MAPI Pharma, MedImmune, Merck/EMD Serono, Novartis, Roche, and Sanofi Genzyme, outside the submitted work. H.W. reports grants and personal fees from AbbVie, Biogen, Merck Serono, and Sanofi Genzyme; personal fees from Actelion, Alexion, Evgen, F. Hoffmann-La Roche, Gemeinnützige Hertie-Stiftung, Immunic, Lundbeck, MedDay Pharmaceuticals, Novartis, Roche Pharma AG, Teva, and WebMD Global; and grants from Deutsche Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, European Union, Fresenius Foundation, German Ministry for Education and Research (BMBF), GlaxoSmithKline, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, NRW Ministry of Education and Research, PML Consortium, RE Children’s Foundation, and Swiss MS Society, outside the submitted work. X.M. reports personal fees and non financial support from Actelion, Biogen, Celgene, Excemed, F. Hoffmann-La Roche, Merck Serono, MSIF, National Multiple Sclerosis Society (NMSS), Novartis, Sanofi Genzyme, and Teva, outside the submitted work. E.A. reports compensation for activities such as advisory boards, lectures, and consultancy from Actelion/Janssen, Alexion, Bayer, Biogen, Celgene/Bristol Myers Squibb, EMD Serono/Merck, Genentech/Roche, Genzyme, Novartis, Sanofi, and TG Therapeutics; and research support from Biogen, Genentech/Roche, National Institutes of Health (NIH), NMSS, Novartis, Patient-Centered Outcomes Research Initiative, Rocky Mountain MS Center, and TG Therapeutics. M.D. reports grants and consulting or speaking fees from Biogen Idec, Celgene/Receptos, Janssen/Actelion, Merck/EMD Serono, Novartis, Roche, and Sanofi Genzyme; S.R.D. reports grants and consulting fees from Novartis; and grants from MAPI Pharma, NIH/NINDS, and NMSS, outside the submitted work. E.P.E. reports grants and consulting or speaking fees from Biogen Idec, Celgene/Receptos, Janssen/Actelion, MedImmune, Merck/EMD Serono, Novartis, Roche, and Sanofi Genzyme. N.G. reports grants and consulting fees from Biogen, Merck Serono, Novartis, Roche, Sanofi Genzyme, and Teva. K.G-P. reports grants and consulting fees from Merck/EMD Serono, Novartis, Roche, Sanofi Genzyme, and Teva. S.H. reports grants and consulting fees from Merck/EMD Serono, Roche, Sanofi Genzyme, and Teva. C.E.H. has nothing to disclose. G.I. reports receiving fees for participating in speaker bureaus and/or advisory boards from Actelion, Bayer, Biogen Idec, Celgene, Merck Serono, Novartis, Roche, Sanofi, and Teva. G.K. has nothing to disclose. F.L. reports grants and personal fees from Biogen and Novartis; and personal fees from Celgene, MedDay, Merck, Roche, Sanofi Genzyme, and Teva. M.M.1 has nothing to disclose. J.E.M-L. reports grants and consulting or speaking fees from Almirall, Biogen, Bristol Myers Squibb, Genzyme, Merck, Novartis, Roche, and Teva. D.M. reports grants and consulting fees from Merck/EMD Serono, Novartis, Roche, and Sanofi Genzyme, outside the submitted work. J.N. reports grants and consulting fees from Alexion, Biogen Idec, Genentech, Genzyme, and Novartis; consulting fees from EMD Serono and Greenwich Biosciences; and speaking fees from Biogen Idec, Consortium of Multiple Sclerosis Centers (CMSC), EMD Serono, Genentech, Novartis, and Viela Bio. D.S.R. reports grant support from Janssen, MedDay Pharmaceuticals, Patient-Centered Outcomes Research Institute, and TG Therapeutics; grants and personal fees from Biogen, EMD Serono, Genentech, Mallinckrodt, Novartis, Prime CME, and Sanofi Genzyme; and personal fees from Alexion, Bristol Myers Squibb, CMSC, and Multiple Sclerosis Association of America. D.V.S. reports personal fees from BIOCAD and Novartis. K.S. reports receiving fees for participating in speakers bureaus from AbbVie and Biohaven Pharmaceuticals. B.S. has nothing to disclose. N.T. reports institutional research fees and personal fees from Actelion/Janssen, Alexion, BIOCAD (Russia), Generium (Russia), MAPI Pharma, Merck, Novartis, Receptos, Roche, Sanofi, and TG Therapeutics. M.V.1 reports speaker fees and consultant fees from Biogen Idec, Merck Serono, Novartis, Roche, Sanofi Genzyme, and Teva. M.V.2 reports compensation for travel, speaker fees, and consultant fees from Biogen Idec, Merck Serono, Novartis, Roche, Sanofi Genzyme, and Teva. M.B., D.A.H., I.L., R.W., M.Z., W.S., and M.M.2 are employees of Novartis Pharma AG. E.J.F. reports compensation for research, consulting, speakers bureau, and/or advisory work from AbbVie, Alexion, Biogen, Bristol Myers Squibb, Chugai, EMD Serono, Genentech/Roche, MedDay, Novartis, Sanofi Genzyme, and TG Therapeutics.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: the APLIOS trial was funded by Novartis Pharmaceuticals. Novartis Pharmaceuticals supported the development of this manuscript, provided data analyses according to the direction of the authors, and paid for medical writing support.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.