
Abstract
Background:
ADS-5102, a delayed-release, extended-release (DR/ER) amantadine, improved walking speed in MS in a Phase 2 trial.
Objective:
The aim of this study was to present primary results of a Phase 3, double-blind, ADS-5102 trial (INROADS) for walking speed.
Methods:
Adult participants with MS and walking impairment, not currently using amantadine or dalfampridine, underwent 4-week placebo run-in before randomization 1:1:1 to placebo, 137 or 274 mg/day ADS-5102 for 12 weeks. Primary outcome was the proportion of responders (20% increase in Timed 25-Foot Walk (T25FW) speed) for 274 mg ADS-5102 versus placebo at end of double-blind (Study Week 16). Additional measures included Timed Up and Go (TUG), 2-Minute Walk Test (2MWT), and 12-item Multiple Sclerosis Walking Scale (MSWS-12).
Results:
In total, 558 participants were randomized and received double-blind treatment. Significantly more participants responded with 274 mg ADS-5102 (21.1%) versus placebo (11.3%). Mean T25FW speed also significantly improved (0.19 ft/s) versus placebo (0.07 ft/s). Other measures were not significant using prespecified hierarchical testing procedure. Adverse events led to discontinuation for 3.8% (placebo), 6.4% (137 mg ADS-5102), and 20.5% (274 mg ADS-5102).
Conclusion:
INROADS met its primary endpoint, showing a significantly greater proportion of participants with meaningful improvement in walking speed for 274 mg ADS-5102 versus placebo. Numeric dose response was seen for some secondary efficacy outcomes and adverse events.
Keywords
Introduction
Multiple sclerosis (MS) is a frequently disabling neurological condition with an estimated worldwide prevalence of 2.1 million people, with about half of these in the United States.1,2 Most individuals (~75%) with MS develop gait and mobility impairments, 3 and half become dependent on walking aids within 15 years of diagnosis.4,5 Although many drugs are approved to reduce relapse rate and/or slow worsening disability in MS,6,7 only extended-release dalfampridine (Ampyra®) in the United States, also known as prolonged-release fampridine (Fampyra®) in other countries, is approved to improve walking in MS, based on demonstrated increases in walking speed during clinical trials.8–12 In addition to treating motor symptoms in Parkinson’s disease, amantadine has been used off-label to treat fatigue in MS and promote wakefulness in other neurologic disorders.13,14 Although best known as a low affinity (Ki 10 µM), uncompetitive N-methyl-D-aspartate (NMDA) receptor inhibitor, at therapeutically relevant doses, amantadine also blocks Kir2 potassium channels.15–17 NMDA receptor inhibition may help normalize aberrant synaptic plasticity in MS, and potassium channel inhibition may help improve nerve conduction across demyelinated regions.
ADS-5102 is a delayed release/extended release (DR/ER) capsule formulation of amantadine designed to be taken once daily at bedtime. Following an initial lag, plasma concentrations rise steadily overnight, providing high daytime concentrations that fall toward evening to minimize impact on sleep. 18 ADS-5102 received FDA approval as Gocovri® (Adamas Pharmaceuticals, LLC., Emeryville, CA) in 2017 to treat levodopa-induced dyskinesia in Parkinson’s disease, and an additional approval in 2021 as a levodopa-adjunct to treat OFF episodes. 19
In a Phase 2, randomized, double-blind, 4-week trial in patients with MS (n = 60), 20 274 mg ADS-5102 improved mean Timed 25-Foot Walk (T25FW)21,22 time by a placebo-adjusted 16.6% and Timed Up and Go (TUG) 23 time by 10%. The drug was generally well tolerated, with dry mouth, constipation, and insomnia as the most common adverse events (AEs). 20 Here, we report results of a subsequent Phase 3 trial, that also included a lower, 137 mg dose, based on the recommendation of regulatory authorities.
Materials and methods
Study design and participants
The Investigational Research Study of ADS-5102 in MS Walking Impairment (INROADS) was a 3-arm, multicenter, randomized, double-blind, 16-week trial (ClinicalTrials.gov NCT03436199) consisting of a screening period; a 4-week single-blind placebo run-in period; and a 12-week double-blind treatment period (Figure 1). The trial was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the principles of the Declaration of Helsinki. Central and local ethics committees approved the trial, and all participants gave written informed consent.
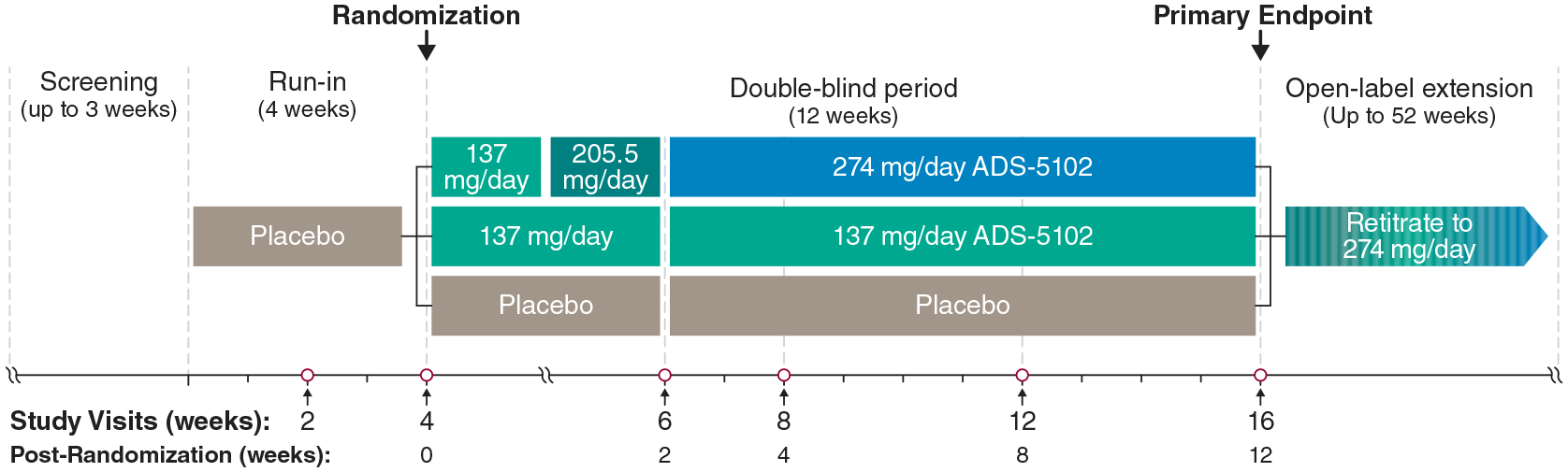
Study design. Doses of 137, 205.5, and 274 mg ADS-5102 (dosed by weight of amantadine base) correspond with 170, 255, and 340 mg amantadine HCl, respectively. Baseline was the average of study visits 2 and 4.
Participants completing the placebo run-in and continuing to meet eligibility criteria were randomized 1:1:1 to placebo or ADS-5102 137 or 274 mg/day, administered as two capsules at bedtime. Participants randomized to the 274 mg/day ADS-5102 dose received 137 mg/day during post-randomization Week 1, 205.5 mg/day during Week 2, and 274 mg/day during post-randomization Weeks 3–12 (Study Weeks 7–16). Participants completing the trial were eligible to enter an optional, single-arm (274 mg ADS-5102) open-label extension trial. Eligible participants were aged 18–70 years, with a diagnosis of MS (McDonald 2017 criteria 24 ) and expanded disability status scale (EDSS) 25 score ⩽ 6.5, were able to complete the T25FW within 8–45 seconds in each of the two trials performed 5 minutes apart at the screening visit, and reported a stable physical activity level and stable MS medications (disease modifying and symptomatic) for 30 days before screening and throughout the study. Subjects using an assistive device during the walking assessments were to use the same assistive device for all subsequent walking tests. Exclusion criteria included participants currently taking amantadine, dalfampridine (including related preparations), or systemic corticosteroids, or onset of a clinically significant MS relapse within 30 days before screening. Additional exclusions included: history of seizures, orthostatic hypotension, suicidal ideation, hallucinations, or other disorders associated with psychosis and estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 (see ClinicalTrials.gov for full list).
Assessments
Efficacy assessments included T25FW, TUG, the 2-Minute Walk Test (2MWT), 26 and the 12-item Multiple Sclerosis Walking Scale (MSWS-12). 27 Safety assessments included AEs, physical examinations, vital signs, electrocardiograms, laboratory measures, and the Columbia-Suicide Severity Rating Scale. 28 Efficacy assessments were performed at Weeks 0, 2, and 4 (the randomization visit) of the placebo run-in, and post-randomization Weeks 4, 8, and 12 of double-blind treatment (Study Weeks 8, 12, and 16; Figure 1). Baseline was defined as the average of Weeks 2 and 4 (just before randomization) of the placebo run-in period. Safety assessments were performed at screening and during each efficacy assessment visit.
Statistical methods
Assuming a placebo response rate ⩽ 20% and active treatment response rate ⩾ 33%, 180 randomized participants per treatment group would be needed to detect a 13% difference in T25FW responder rate favoring either active treatment dose versus placebo (Farrington–Manning approach for the Miettinen–Nurminen test) to have 80% power at the two-sided 5% significance level. Assuming 5% dropout during placebo run-in, enrollment of 570 participants was needed to randomize 540 participants.
The efficacy analysis for the primary and key secondary endpoints used the intent-to-treat (ITT) population, defined as all randomized participants who received a dose of double-blind treatment. The primary efficacy analysis was the treatment difference (ADS-5102 minus placebo) in the proportion of responders at 12 weeks post randomization (Study Week 16), for the 274 mg ADS-5102 group. Response was defined as a ⩾ 20% increase in walking speed (average ft/s during two repetitions of the T25FW) from baseline. Walking speed ft/s was used for T25FW since walking speed is more normally distributed as compared to walking time, and is therefore a preferred approach.29,30 A 20% change in T25FW is considered a meaningful change in patients with MS.21,22,29,31,32 Participants missing the final double-blind assessments (Study Week 16) were categorized as nonresponders. Superiority was to be concluded if the two-sided p value, obtained using the Farrington–Manning test, was less than 0.05 and the lower limit of the two-sided 95% CI for the treatment difference (ADS-5102 274 mg minus placebo; obtained using the Miettinen–Nurminen approach) was greater than 0. Provided the primary analysis demonstrated superiority, the key secondary outcomes were sequentially evaluated, in hierarchical order, using a fixed-sequence gatekeeping strategy to control the overall type 1 (false positive) error rate at 5%. Analyses were performed in the following sequence: T25FW responder rate and then change from baseline in T25FW, TUG, and 2MWT for the 274 mg dose versus placebo, followed by responder rate and change from baseline in each of these efficacy measures for the 137 mg dose versus placebo. At each step, if superiority was not demonstrated, all subsequent results were considered statistically nonsignificant, irrespective of p-value.
Hypothesis testing for the T25FW, TUG, and 2MWT was done for both ADS-5102 doses and placebo using t-tests derived from the corresponding linear mixed model with repeated measures (MMRM) model, with the change from baseline as the dependent variable and fixed effects of treatment group, study week, and treatment by study week interaction. The baseline value was included as a covariate, and an unstructured variance–covariance matrix was used for the within-participant residual variability. If the model failed to converge under this assumption, then a compound symmetry covariance structure was assumed. The Kenward–Roger method was used to estimate the denominator degrees of freedom. Prespecified analyses to assess the impact of missing data included repeating the primary analysis using study completers. Where permitted by sample size (at least 30 per group), the primary responder analysis was also repeated for prespecified subgroups based on baseline demographic and disease state categories (age, gender, race, BMI, type of MS, time since MS diagnosis, EDSS score, use of assistive devices, history of dalfampridine or amantadine use, and T25FW time).
Treatment-emergent AEs (TEAEs) were coded and summarized according to the Medical Dictionary for Regulatory Activities (version 21.0). AEs recorded during single-blind placebo treatment were not included in reporting. Software used was SAS version 9.4 (SAS Institute Inc., Cary, NC).
Role of the funding source
The sponsor contributed to study design, conduct, and reporting. External authors had full access to study data, participated in the planning of the publication, and had final responsibility for the decision to submit the paper for publication.
Results
Study population
The study was performed at 85 centers in the United States and Canada between April 2018 and November 2019. Disposition for the 594 enrolled participants is shown in Figure 2. A total of 558 randomized participants received double-blind treatment with placebo (n = 186), 137 mg ADS-5102 (n = 187), or 274 mg ADS-5102 (n = 185) and composed the ITT and safety populations. Of these, 473 (placebo (n = 174; 93.5%), ADS-5102 137 mg (n = 166; 88.8%), and ADS-5102 274 mg (133; 71.9%)) completed study drug treatment. Of the 85 participants who discontinued study drug, 11 (placebo (n=2), ADS-5102 137 mg (n=3), and ADS-5102 274 mg (n=6)) remained in the study and continued to undergo assessments. Two patients (n = 1 in each ADS-5102 group) completed treatment (Week 16) but did not complete the safety follow-up visit and were therefore listed as not completing the study.
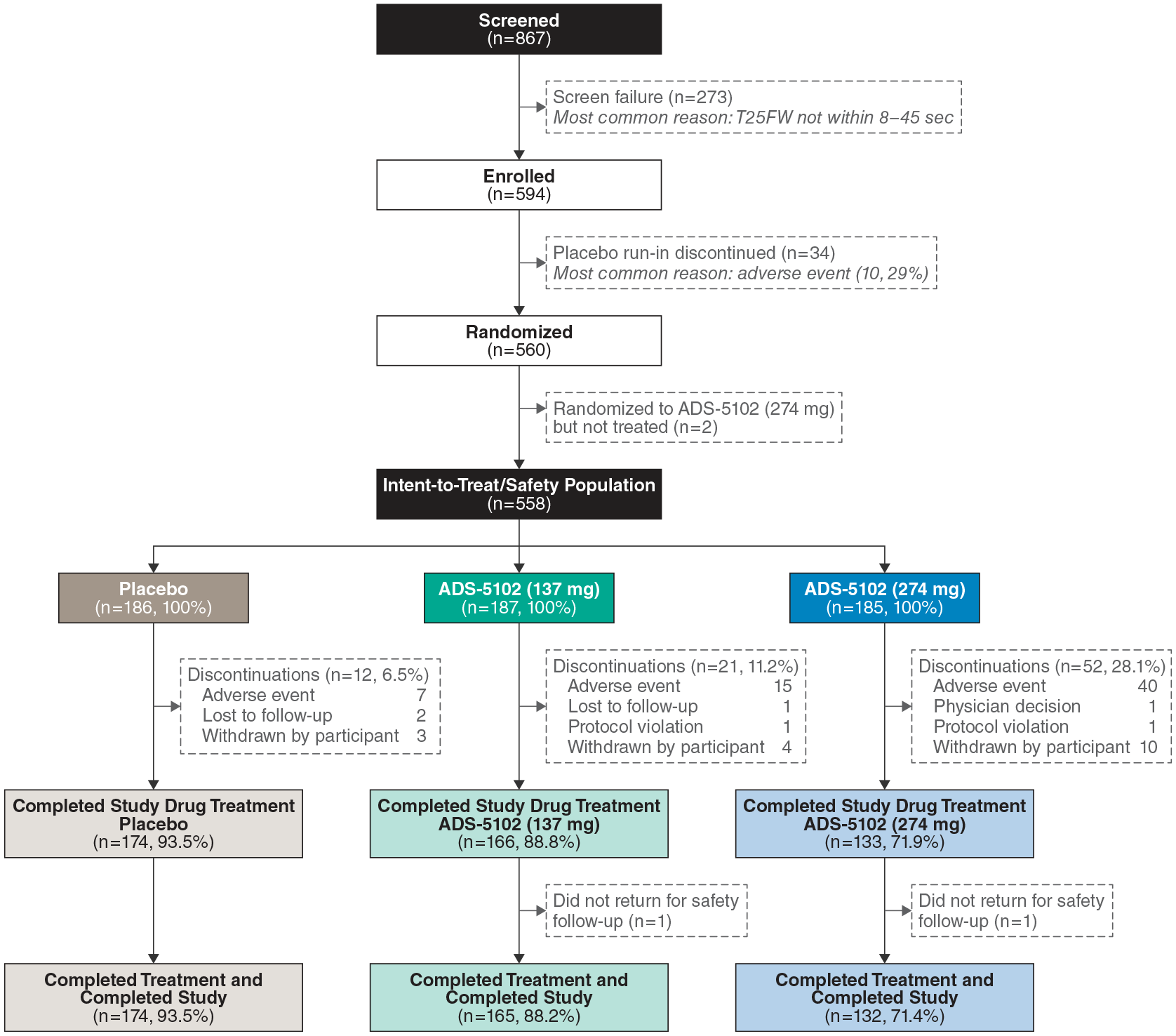
Participant disposition. A total of 11 participants (2 (1.1%) placebo, 3 (1.6%) ADS-5102 137 mg, and 6 (3.2%) ADS-5102 274 mg) who discontinued treatment before Week 16 continued to receive assessments in the study. Therefore, a total of n = 176 (placebo), n = 168 (ADS-5102 137 mg), and n = 138 (ADS 5102 274 mg) participants, respectively, completed the Week 16 study assessments. Two patients (one in each ADS-5102 group) subsequently did not return for the safety follow-up visit and were counted as having completing study treatment, but not as completing the study.
Baseline characteristics are shown in Table 1. Overall, participants had a mean age of 54.4 years, with a majority being ⩾ 55 years of age, female, and white. Most participants (95.5%) performed the T25FW in < 26.5 seconds. At screening, approximately half of participants (55.6%) had an EDSS score of 6.0 or 6.5. In total, 72 (12.9%) participants had previously used amantadine and 293 (52.5%) had previously used dalfampridine (distribution by treatment group shown in Table 1); 72% were concomitantly using disease-modifying treatments for MS, including glatiramer, beta-interferons, alemtuzumab, fingolimod, natalizumab, ocrelizumab, teriflunomide, or dimethyl fumarate.
Baseline demographics, disease characteristics, and walking assessments.
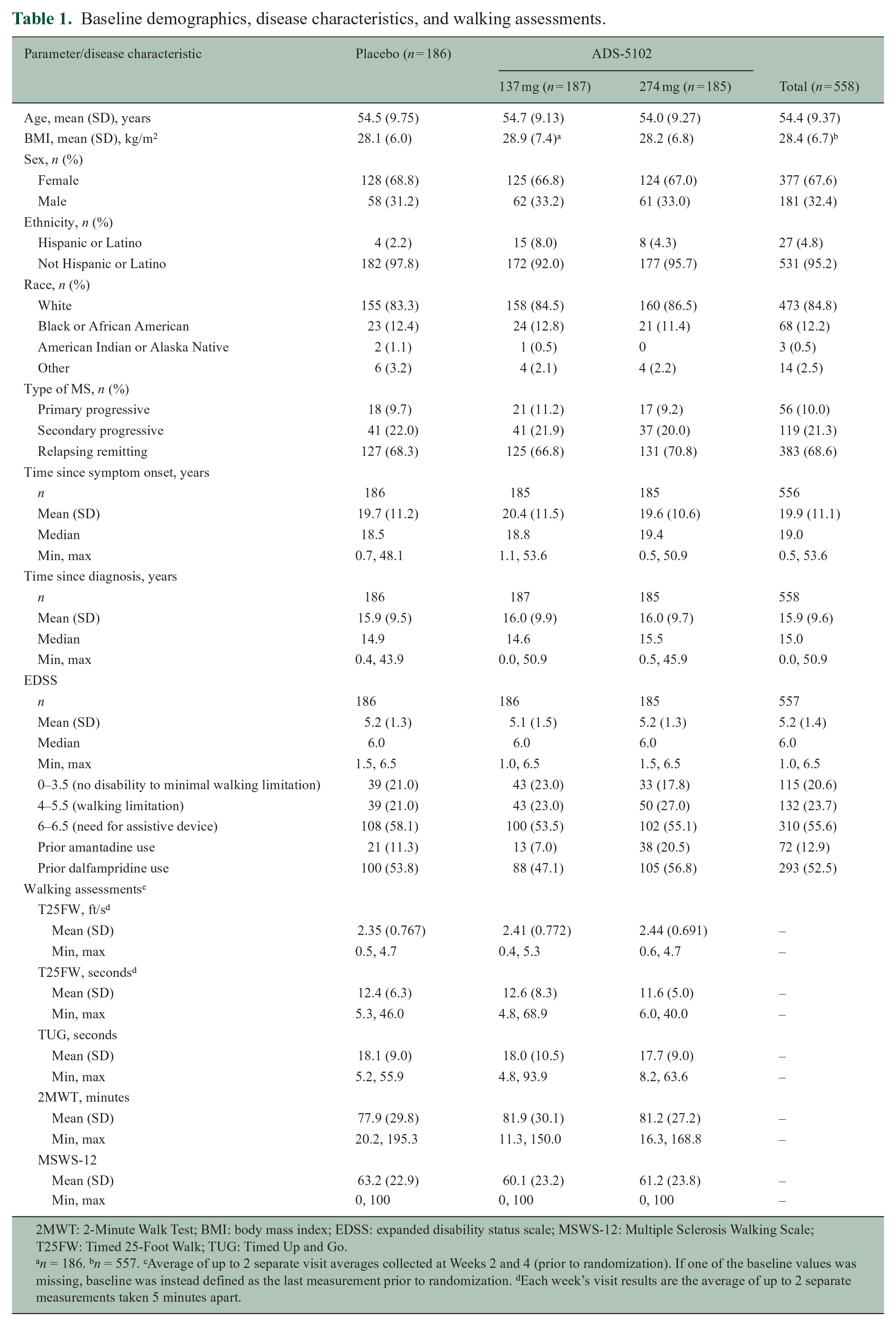
2MWT: 2-Minute Walk Test; BMI: body mass index; EDSS: expanded disability status scale; MSWS-12: Multiple Sclerosis Walking Scale; T25FW: Timed 25-Foot Walk; TUG: Timed Up and Go.
n = 186. bn = 557. cAverage of up to 2 separate visit averages collected at Weeks 2 and 4 (prior to randomization). If one of the baseline values was missing, baseline was instead defined as the last measurement prior to randomization. dEach week’s visit results are the average of up to 2 separate measurements taken 5 minutes apart.
Efficacy
The proportion of participants meeting responder criteria (⩾ 20% increase in T25FW speed) was 21.1% for 274 mg ADS-5102 (n/N = 39/185) and 11.3% for placebo (n/N = 21/186), representing a significant risk difference of 0.098 (95% CI: 0.023–0.174; p = 0.01) (Figure 3(a)). This significant treatment effect was maintained when restricting the analysis to study completers: the proportion of responders was 28.3% for 274 mg ADS-5102 (39 of 138 completers) and 11.9% for placebo (21 of 176 completers), representing a risk difference of 0.163 (95% CI: 0.075–0.254; p < 0.001) (Figure 3(b)). The cumulative distribution of T25FW response at Week 12 post randomization is shown in Figure 3(c). The difference between 274 mg ADS-5102 and placebo was significant only for the ⩾ 20% responder threshold (p = 0.01). Separation from placebo in T25FW speed was present by post-randomization Week 4 and widened by post-randomization Week 12 (Study Week 16) (Figure 3(d)).
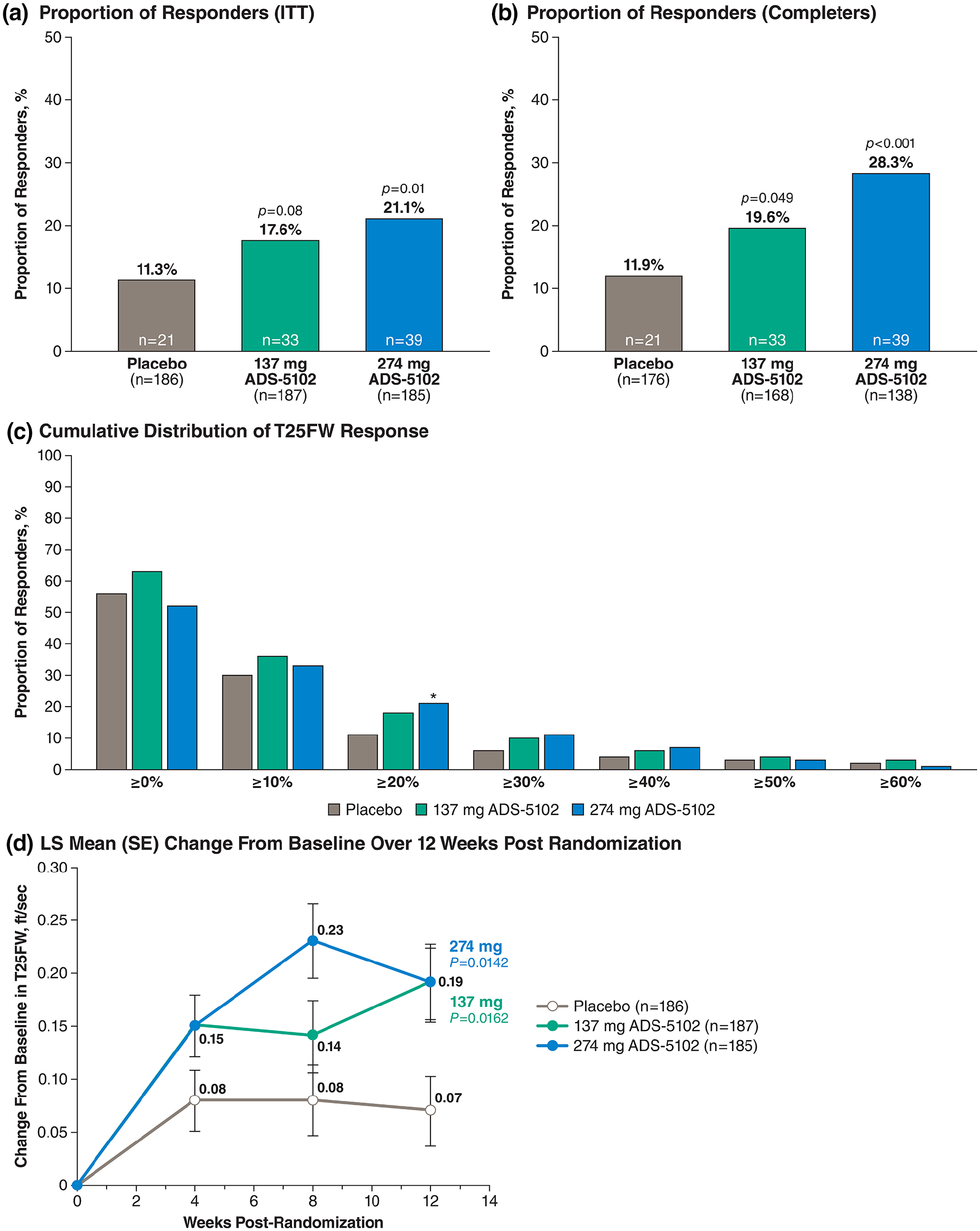
T25FW responder analyses and mean change at 12 weeks post randomization (Study Week 16). (a) Proportion of responders (denominators are ITT subjects). (b) Proportion of responders (denominators are subjects completing week 16). (c) Cumulative distribution of T25FW response (denominators are ITT subjects). (d) LS mean (SE) T25FW change from baseline over 12 weeks post randomization (Study Week 16).
Prespecified subgroup analyses generally supported the primary analysis. The proportion of participants meeting T25FW responder criteria was similar irrespective of prior dalfampridine use, with respective percentages for former dalfampridine users and nonusers of 20% and 22.5% for 274 mg ADS-5102, and 11.0% and 11.6% for placebo.
Forest plots for prespecified responder rate subanalyses at post-randomization Week 12 are shown in Figure 4. Subpopulations with significant risk differences favoring 274 mg ADS-5102 over placebo included age < 55 years (p = 0.003), female sex (p = 0.016), lower time since diagnosis (p = 0.039), not using an assistive device (p = 0.025), and EDSS score 4–5.5 (p = 0.009). Analysis of secondary endpoints at post-randomization Week 12 showed statistical significance for 274 mg ADS-5102 over placebo for change in walking speed (T25FW, least squares mean treatment difference, 0.12 ft/s; p = 0.01; Table 2), but not for TUG test results (least squares mean treatment difference, −0.4, p = 0.95); as such, the result for the 2MWT, despite returning a p-value < 0.05 must be considered not significant based on the hierarchical testing procedure. The responder rate for the 137 mg ADS-5102 dose was 17.6% (n/N = 33/187), representing a risk difference of 0.064 (95% CI: −0.008–0.136; p = 0.08). Although the mean change in T25FW speed and 2MWT for 137 mg ADS-5102 returned p-values < 0.05, these also must be considered not significant. Of note, the 2MWT results showed a rising trend for ADS-5102, with non-overlap of CIs by Week 4 post randomization (Supplementary Figure 1). Mean changes in MSWS-12 score were small and similar across treatment groups.
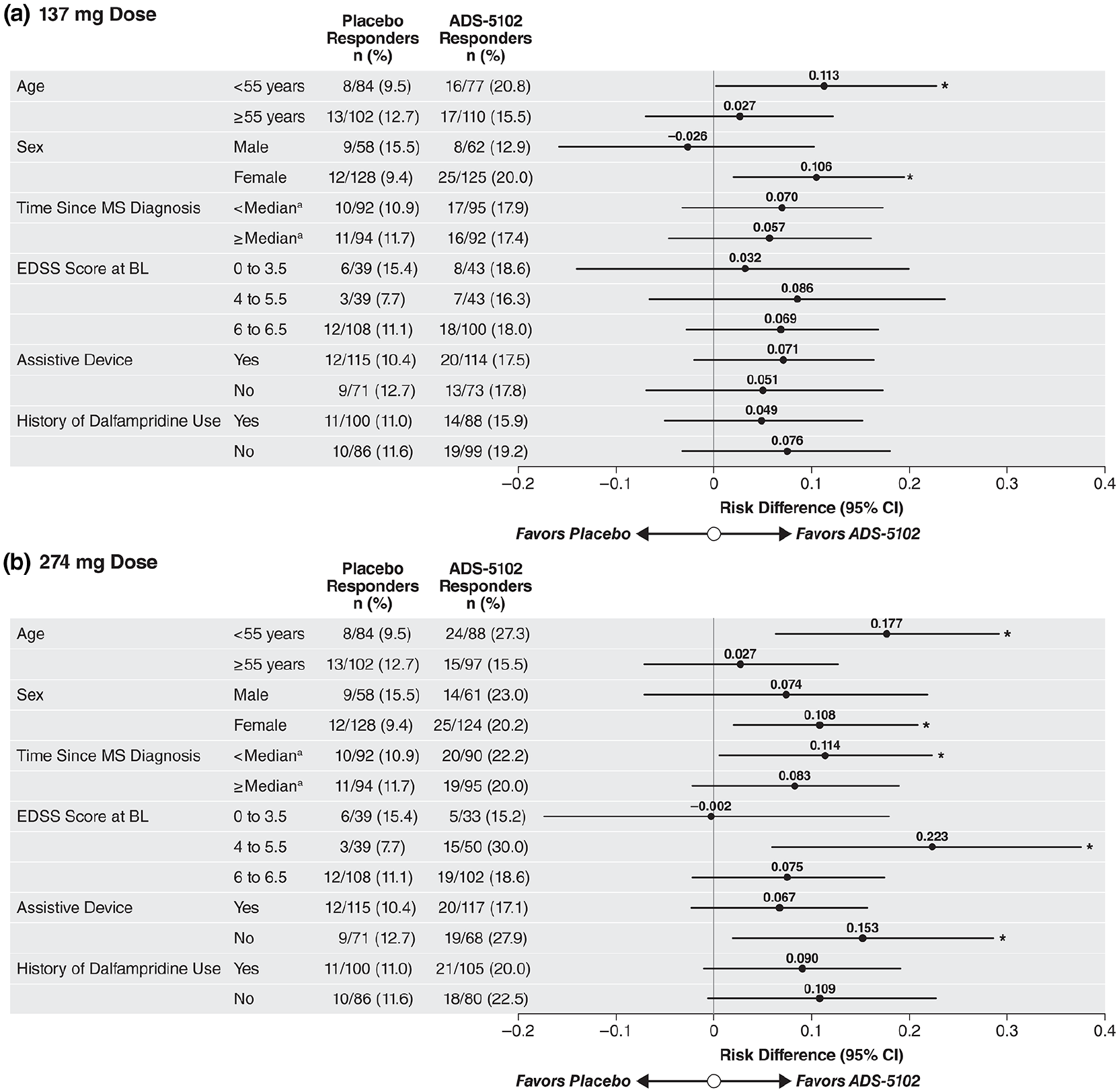
T25FW responder analysis by prespecified subgroups at 12 weeks post randomization (Study Week 16) based on baseline demographic and disease-state categories. (a) 137 mg dose. (b) 274 mg dose.
Mean change from baseline to Week 12 post randomization (Study Week 16) in the T25FW, TUG, 2MWT, and MSWS-12 (ITT population).

2MWT: 2-Minute Walk Test; CI: confidence interval; LS: least squares; MMRM: mixed model with repeated measures; MSWS-12: 12-item Multiple Sclerosis Walking Scale; T25FW: Timed 25-Foot Walk; TUG: Timed Up and Go.
p values were based on the comparison between ADS-5102 treatment and placebo from the MMRM model with change from baseline as the dependent variable and the baseline value as a covariate. The model included fixed effects for treatment group, visit (Weeks 4, 8, and 12 post randomization), and the interaction between treatment group and visit. An unstructured covariate structure was used.
Statistically significant based on the hierarchical strategy.
MSWS-12 MMRM results were calculated post hoc.
A post hoc analysis by responder status for all efficacy outcomes is shown in Figure 5.
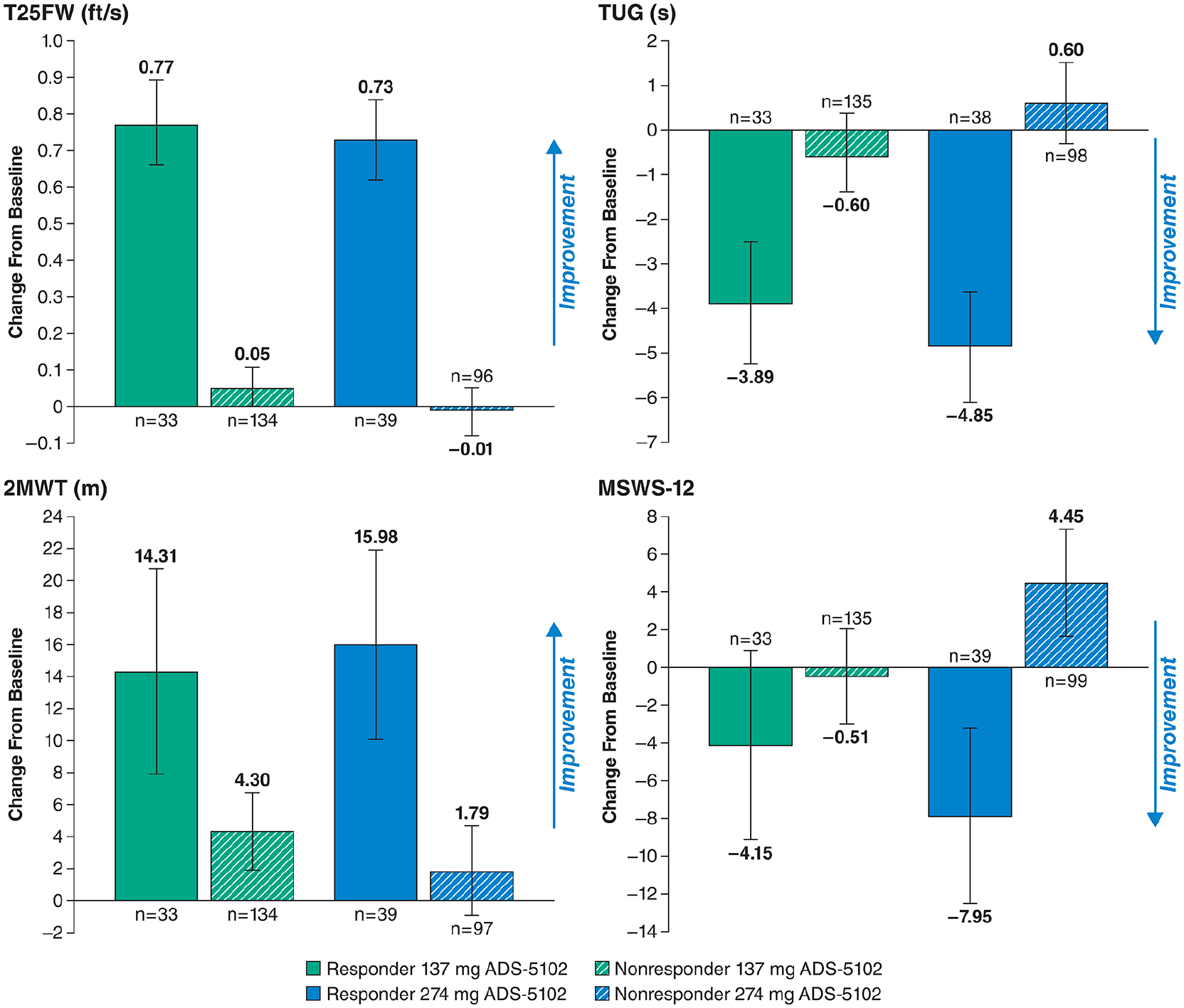
Analysis (MMRM) by responder status of efficacy endpoint changes from baseline to 12 weeks post randomization (Study Week 16); changes shown are LS mean (95% CI) from the MMRM model with change from baseline as the dependent variable and the baseline value as a covariate. The model was run separately for responders and nonresponders, and included fixed effects for treatment group, visits (Weeks 8, 12, and 16), and the interaction between treatment group and visit. An unstructured covariate structure is used. 2MWT, 2-Minute Walk Test; MMRM, mixed model with repeated measures; MSWS-12, 12-item Multiple Sclerosis Walking Scale; T25FW, Timed 25-Foot Walk; TUG, Timed Up and Go.
Safety
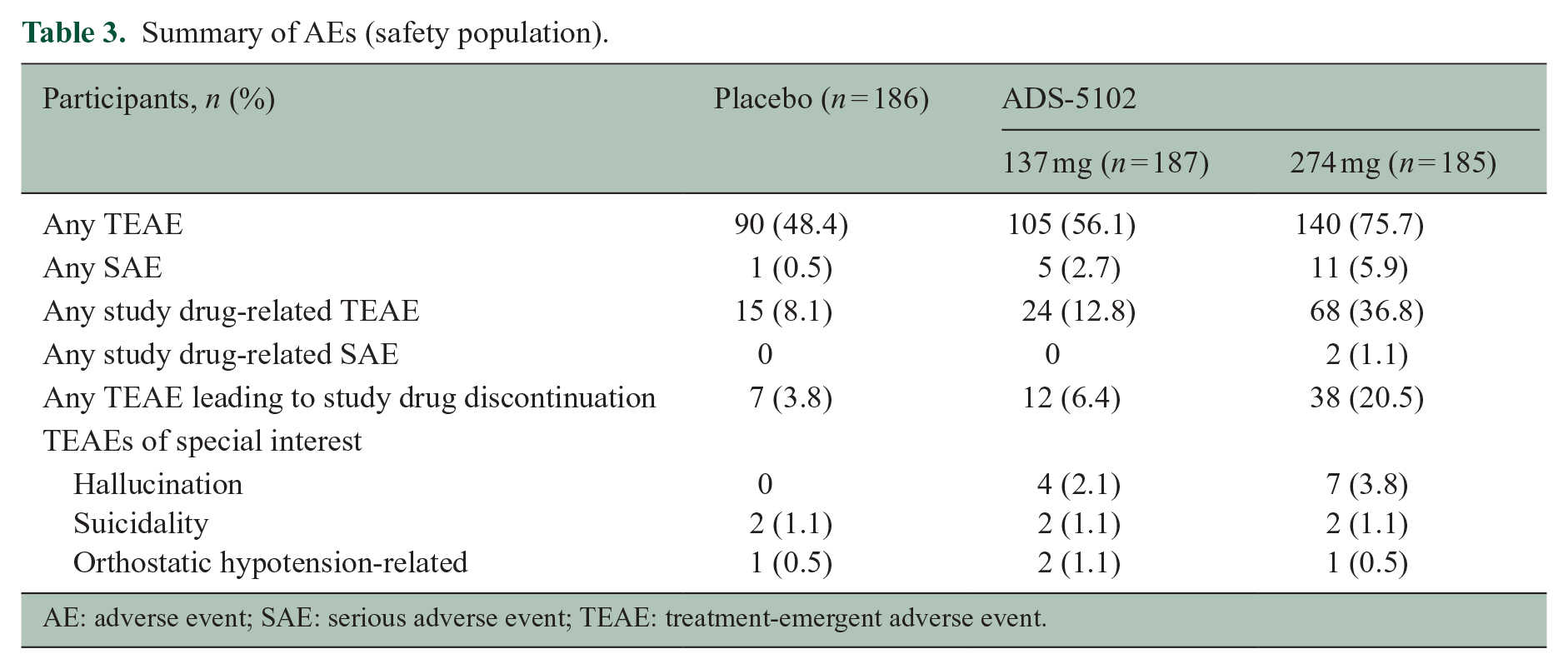
Table 3 summarizes AEs, and the most commonly reported TEAEs and treatment discontinuation rate are displayed in Table 4. Two events, peripheral edema and dry mouth, occurred in ⩾ 10% of ADS-5102-treated patients in either dose group. Peripheral edema was considered related to study drug for 0.5%, 3.2%, and 10.8% randomized to placebo, 137 mg ADS-5102, and 274 mg ADS-5102, respectively, and dry mouth was considered related for 1.1%, 1.1%, and 9.7%, respectively. AEs were typically mild or moderate in severity. Severe TEAEs were reported for 2.2% receiving placebo, 4.3% receiving 137 mg ADS-5102, and 8.1% receiving 274 mg ADS-5102. Events rated as severe for > 1 ADS-5102-treated participant were peripheral edema (n = 2), appendicitis (n = 2), and insomnia (n = 2). AEs led to study drug discontinuation for 3.8%, 6.4%, and 20.5% in the placebo, 137 mg ADS-5102, and 274 mg ADS-5102 groups, respectively, most frequently due to peripheral edema, visual hallucinations, insomnia, tremor, or dry mouth.
Summary of AEs (safety population).
AE: adverse event; SAE: serious adverse event; TEAE: treatment-emergent adverse event.
Incidence of common TEAEs (⩾ 3% in either active arm) and number of patients discontinuing study treatment for these events.
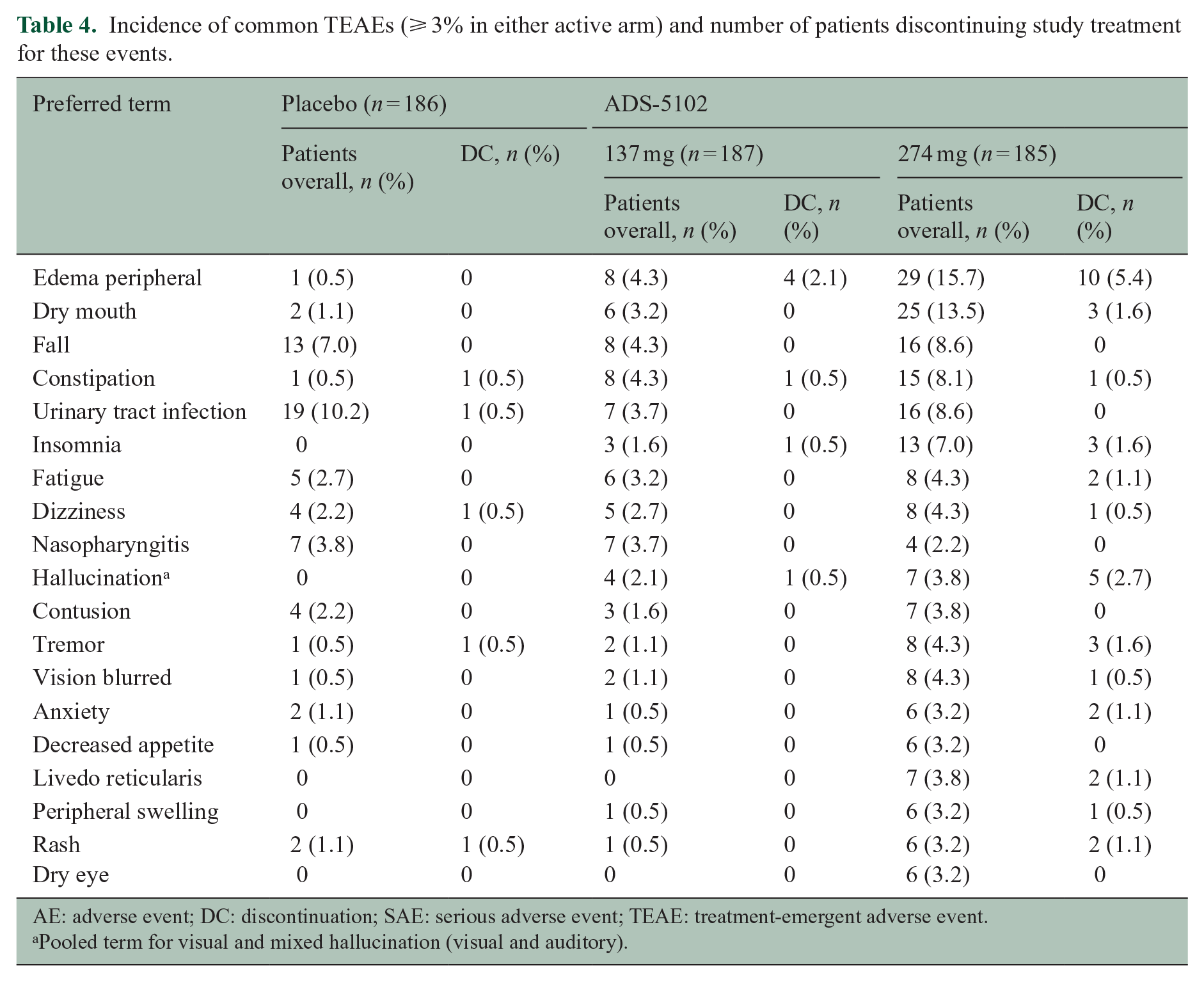
AE: adverse event; DC: discontinuation; SAE: serious adverse event; TEAE: treatment-emergent adverse event.
Pooled term for visual and mixed hallucination (visual and auditory).
Serious AEs (SAEs) were reported for 1 (0.5%), 5 (2.7%), and 11 (5.9%) participants receiving placebo, 137 mg-, or 274 mg ADS-5102, respectively. The only SAEs occurring in > 1 ADS-5102-treated participant were MS relapse (n = 2 receiving 274 mg) and appendicitis (n = 1 each ADS-5102 dose). SAEs led to discontinuation for six patients (all receiving ADS-5102) and included acute myocardial infarction (n = 1 (137 mg)) and hallucinations, ileus, osteoarthritis, renal cell carcinoma, and blurred vision (n = 1 each (274 mg)). All SAEs, except the reports of hallucination and blurred vision that led to discontinuation, were considered unrelated to ADS-5102 treatment. No deaths occurred during the study. In addition to hallucinations (reported above and in Table 3), suicidality, and orthostatic hypotension were prespecified as events of special interest. Orthostatic hypotension-like events were reported for 0.5%, 1.1%, and 0.5% of participants receiving placebo, 137 mg ADS-5102, and 274 mg ADS-5102, respectively, and suicidality was reported as an AE for 1.1% in each treatment group.
No clinically relevant vital sign changes or physical examination findings were noted. Four participants had potentially clinically significant (PCS) laboratory values that were also reported as TEAEs: leukopenia (n = 1 receiving placebo) and liver function test (LFT) elevation (n = 3 randomized to 274 mg ADS-5102). Of those with LFT elevation, one case (considered unrelated to treatment) was reported 23 days after the last dose of ADS-5102. The other two participants (considered treatment related) were taking medications known to cause LFT elevation (nitrofurantoin in both, and teriflunomide in one); PCS LFT elevation (alkaline phosphatase and gamma-glutamyl transferase) resolved in one participant while still on ADS-5102, but off nitrofurantoin; in the other participant, ADS-5102 had been stopped 11 days before LFT elevations (alanine aminotransferase and gamma-glutamyl transferase) were first observed and resolved approximately 2 months after stopping teriflunomide and administering cholestyramine.
Discussion
This Phase 3, double-blind, randomized, placebo-controlled trial met the primary endpoint, with a significantly greater proportion of participants receiving 274 mg ADS-5102 showing a ⩾ 20% increase in T25FW speed at post-randomization Week 12 versus placebo. The 274 mg ADS-5102 group also had significantly improved T25FW speed versus placebo. Although mean changes in T25FW speed and 2MWT favored 137 mg ADS-5102 over placebo, p-values cannot be interpreted as significant based on hierarchical testing procedure. The proportion T25FW responders appeared similar, irrespective of prior dalfampridine use. The rationale for using responder analysis as the primary endpoint, in addition to the proven validity and regulatory acceptability of this approach, was that, similar to dalfampridine and other symptomatic treatments, we anticipated some patients would benefit from ADS-5102 treatment and some may not. The 17.6% (137 mg) and 21.1% (274 mg) responder rates for ADS-5102 (vs 11.3% placebo) were lower than those for the two Phase 3 studies of dalfampridine where ~33% of participants had a ⩾ 20% T25FW speed improvement versus 13.7% for placebo after 9–14 weeks treatment. 33 The response rate for 274 mg ADS-5102 was affected by tolerability, as demonstrated by the higher, 28.3% response rate in completers.
Although significant, the overall improvement in T25FW (0.12 ft/s over placebo) and the low magnitude of treatment differences for other secondary endpoints may seem of questionable clinical relevance. However, it is important to keep in mind that our prespecified analyses were conducted by treatment group (study drug vs placebo) instead of by responder status (T25FW responders vs nonresponders) within treatment arms as was done in the dalfampridine trials.11,12,33 A similarly conducted post hoc analysis by responder status for our study shows the validity of the responder definition, with demonstrable differences for all efficacy outcomes (Figure 5). For example, the least squares mean change in T25FW for 274 mg ADS-5102 responders is 0.73 ft/s, which is above the 0.36 ft/s minimal clinically important difference (MCID) reported by Coleman et al in 2012, 34 and greater than the −0.1 ft/s change for nonresponders. Similarly, evaluation of 2MWT shows results of 14.3 (137 mg) and 16.0 (274 mg) for responders (vs 3.4 and 1.8, respectively, for nonresponders), clearly exceeding the MCID thresholds calculated in an MS population by Baert et al that demonstrated meaningful changes from a patient and therapist perspective of 9.6 m and 6.8 m, respectively. 35 Likewise, the MSWS-12 showed a greater than 12-point treatment difference for 274 mg ADS-5102 (−7.95 point improvement for responders vs + 4.45 point decline for nonresponders) and a 3.6 point difference for 137 mg (−4.15 vs −0.53 improvements, respectively).
Although the 274 mg ADS-5102 dose showed a greater response rate than the 137 mg dose, it also showed poorer tolerability, with higher incidence of drug-related AEs (37% vs 13%, respectively), discontinuations due to AEs (20.5% vs 6.4%), and overall study dropout (25% vs 10%) that may affect drug utility for some patients. Acknowledging these tolerability differences, AEs at both doses were largely assessed as mild or moderate in severity, with peripheral edema or dry mouth being most common. Overall, AEs were consistent with the known safety profile of amantadine.
The fact that we did not control for medication regimen differences across groups is a potential limitation of the study, although subgroup analysis did show a largely consistent response. It is also unknown how effects may change over a longer duration of time than that evaluated in this 16-week study.
In conclusion, the INROADS trial met its primary endpoint, demonstrating that a higher proportion of participants achieved a clinically meaningful improvement in walking speed for 274 mg ADS-5102 compared with placebo. This dose achieved a modestly greater response compared with the 137 mg dose; however, AEs and tolerability also appeared dose-related. Although overall responder rates were not large, the results suggest a role for 274 mg ADS-5102 to improve walking in certain patients with MS. Further insights and safety data will be gained from the ongoing 1-year open-label extension to this trial.
Supplemental Material
sj-pdf-1-msj-10.1177_13524585211035333 – Supplemental material for A Phase 3, double-blind, placebo-controlled efficacy and safety study of ADS-5102 (Amantadine) extended-release capsules in people with multiple sclerosis and walking impairment
Supplemental material, sj-pdf-1-msj-10.1177_13524585211035333 for A Phase 3, double-blind, placebo-controlled efficacy and safety study of ADS-5102 (Amantadine) extended-release capsules in people with multiple sclerosis and walking impairment by Jeffrey A Cohen, Michelle H Cameron, Myla D Goldman, Andrew D Goodman, Aaron E Miller, Anne Rollins, Lily Llorens, Rajiv Patni, Robert Elfont and Reed Johnson in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors thank the INROADS investigators, study staff, and patients for their contributions to the research presented in this paper. Editorial assistance was provided by Robin Smith, PhD, of The Curry Rockefeller Group, LLC (Tarrytown, NY), and was funded by Adamas Pharmaceuticals, Inc. Andrea E. Formella, PharmD, and Judy Lytle, PhD, provided editorial assistance, data QC, and project management. Rob Howard of Veridical Solutions provided data analysis services.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.A.C. has received personal compensation for consulting for Adamas, Atara, Bristol Myers Squibb, Convelo, MedDay, and Mylan and for serving as an editor of Multiple Sclerosis Journal. M.H.C. has received personal compensation as a consultant for Adamas, Greenwich Biosciences, MedLink, and Helius Medical Technologies. M.D.G. has served as a consultant for Adamas, Biogen Idec, Brainstorm Cell Therapeutics Ltd., EMD Serono, Genentech, Greenwich Biosciences, Immunic, MedDay, and Sanofi Genzyme; she is funded by National MS Society Grant RG-2004-36580. A.D.G. has received consulting fees from Adamas, Greenwich Biosciences, and SwanBio; his employer has received research support from Biogen and Sanofi Genzyme. A.E.M. has served as a consultant for AbbVie, Caremark, Adamas, Biogen Idec, Bristol Myers Squibb/Celgene, Corrona, EMD Serono, Mapi-Pharma, Mylan, Novartis, and Roche/Genentech; he has received research support from Genzyme/Sanofi, Novartis, Roche/Genentech, and MedDay, and is on the speaker’s bureau (providing unbranded programs only) at EMD Serono (journal club), Alexion, Biogen Idec, and Genentech (disease awareness programs). A.R. and R.J. are employees and own stock in Adamas Pharmaceuticals, Inc. L.L. is a former employee of, and currently receives personal compensation as a consultant for Adamas Pharmaceuticals. R.P. and R.E. are former employees of and own stock in Adamas Pharmaceuticals.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data availability
The datasets generated during and/or analyzed during the current study are available from Adamas Pharmaceuticals on reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.