
Abstract
Background:
The envelope protein of human endogenous retrovirus W (HERV-W-Env) is expressed by macrophages and microglia, mediating axonal damage in chronic active MS lesions.
Objective and Methods:
This phase 2, double-blind, 48-week trial in relapsing-remitting MS with 48-week extension phase assessed the efficacy and safety of temelimab; a monoclonal antibody neutralizing HERV-W-Env. The primary endpoint was the reduction of cumulative gadolinium-enhancing T1-lesions in brain magnetic resonance imaging (MRI) scans at week 24. Additional endpoints included numbers of T2 and T1-hypointense lesions, magnetization transfer ratio, and brain atrophy. In total, 270 participants were randomized to receive monthly intravenous temelimab (6, 12, or 18 mg/kg) or placebo for 24 weeks; at week 24 placebo-treated participants were re-randomized to treatment groups.
Results:
The primary endpoint was not met. At week 48, participants treated with 18 mg/kg temelimab had fewer new T1-hypointense lesions (p = 0.014) and showed consistent, however statistically non-significant, reductions in brain atrophy and magnetization transfer ratio decrease, as compared with the placebo/comparator group. These latter two trends were sustained over 96 weeks. No safety issues emerged.
Conclusion:
Temelimab failed to show an effect on features of acute inflammation but demonstrated preliminary radiological signs of possible anti-neurodegenerative effects. Current data support the development of temelimab for progressive MS.
Trial registration:
CHANGE-MS: ClinicalTrials.gov: NCT02782858, EudraCT: 2015-004059-29; ANGEL-MS: ClinicalTrials.gov: NCT03239860, EudraCT: 2016-004935-18
Introduction
Human endogenous retroviruses (HERVs) are retroviral germline DNA insertions, accounting for approximately 8% of the human genome. 1 While most HERVs are transcriptionally silent, transcriptional and protein expression can occur and may be involved in the pathogenesis of neurological and other diseases. 2 In multiple sclerosis (MS), the envelope protein (ENV) encoded by a member of the HERV-W family has been suggested to play a pathogenic role, as it is expressed in the brain, most prominently by macrophages and microglia in chronic active lesions.1,3–6 In vitro, HERV-W-ENV induces release of pro-inflammatory cytokines by peripheral blood mononuclear cells, maturation of dendritic cells, and a pro-inflammatory phenotype of microglia leading to axonal damage. HERV-W-ENV also inhibits oligodendrocyte precursor cell (OPC) differentiation.4,5,7–10
Temelimab (formerly GNbAC1) is a humanized IgG4 monoclonal antibody that binds HERV-W-ENV and antagonizes its capacity for immune cell activation and inhibition of OPC maturation.7,11 Therefore, we hypothesized that functional neutralization of HERV-W-ENV could be a novel therapeutic approach in MS.
The phase 2b CHANGE-MS study was designed to evaluate the efficacy and safety of temelimab in patients with relapsing-remitting MS (RRMS). Assuming an anti-inflammatory mode of action, a conventional trial design based on acute gadolinium-enhancing (GdE) lesion formation as the primary endpoint was used. 12 Secondary endpoints related to neurodegeneration such as brain atrophy, number and size of T1 hypointense lesions, and myelin integrity. Here, we report results from the 48-week core study (CHANGE-MS) and subsequent 48-week extension study (ANGEL-MS).
Methods
Participants
Adults aged 18–55 years, weighing 40–100 kg, with RRMS according to 2010 McDonald criteria 13 were included. Participants had to exhibit MS disease activity with ⩾1 relapse within 1 year, or 1 GdE T1 lesion within ⩽3 months, show clinical stability for ⩽30 days before screening, and have an Expanded Disability Status Scale (EDSS) score < 6.0 at study entry. Exclusion criteria included history/diagnosis of primary/secondary progressive MS, complete transverse myelitis or bilateral optic neuritis, use of corticosteroids, or adrenocorticotropic hormone ⩽ 30 days prior to screening, and grade ⩾ 2 lymphopenia following immunosuppressive/immunomodulatory treatment.
Study design
Conducted between May 2016 and November 2018, CHANGE-MS (core study) was a phase 2b, double-blind, randomized, placebo-controlled, parallel-group, multicenter (see Supplementary Table 1) study, 14 followed by an extension study. The extension, ANGEL-MS, was prematurely terminated after 1 year due to withdrawal of funding by the development partner.
Secondary efficacy endpoints.
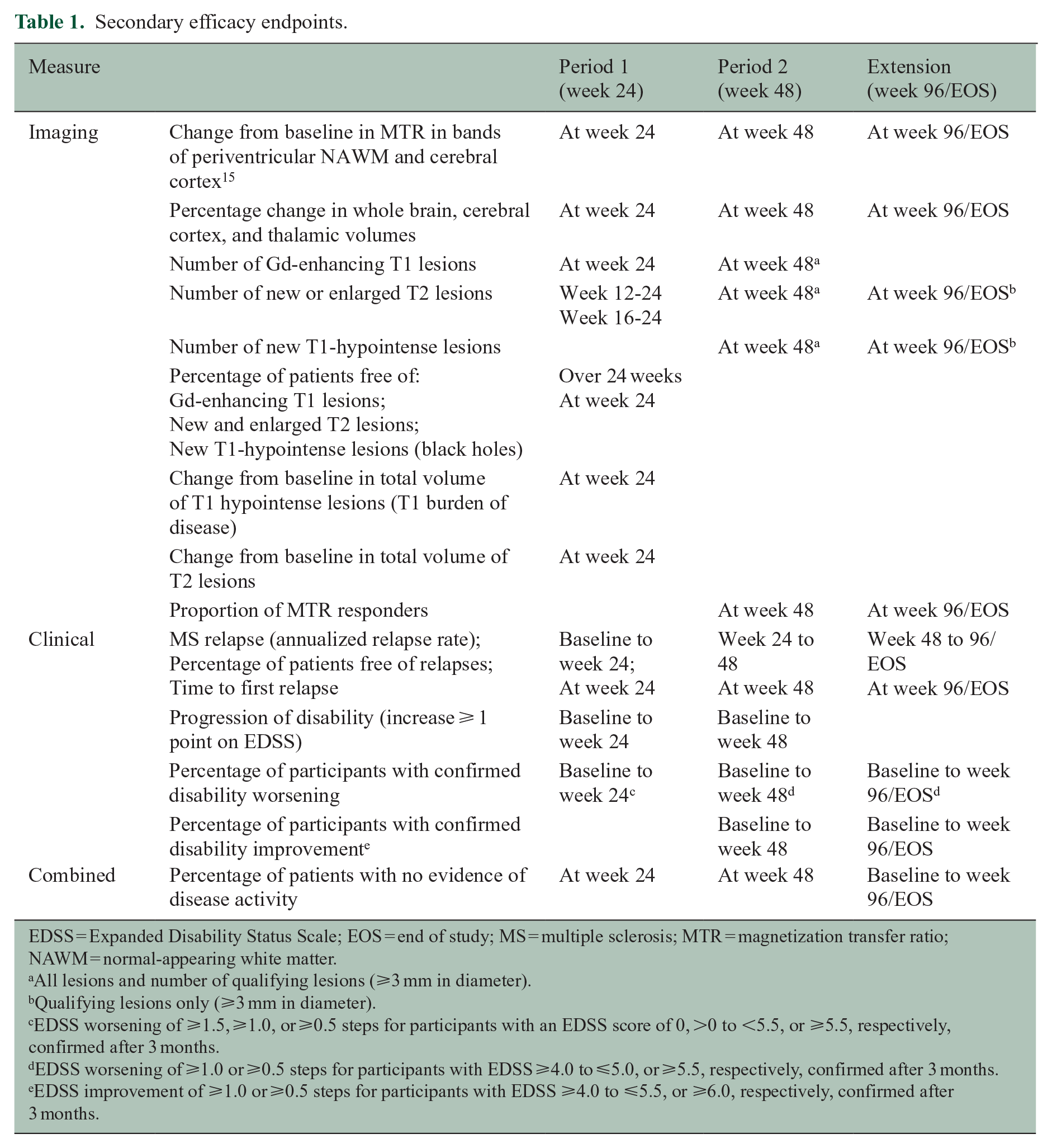
EDSS = Expanded Disability Status Scale; EOS = end of study; MS = multiple sclerosis; MTR = magnetization transfer ratio; NAWM = normal-appearing white matter.
All lesions and number of qualifying lesions (⩾3 mm in diameter).
Qualifying lesions only (⩾3 mm in diameter).
EDSS worsening of ⩾1.5, ⩾1.0, or ⩾0.5 steps for participants with an EDSS score of 0, >0 to <5.5, or ⩾5.5, respectively, confirmed after 3 months.
EDSS worsening of ⩾1.0 or ⩾0.5 steps for participants with EDSS ⩾4.0 to ⩽5.0, or ⩾5.5, respectively, confirmed after 3 months.
EDSS improvement of ⩾1.0 or ⩾0.5 steps for participants with EDSS ⩾4.0 to ⩽5.5, or ⩾6.0, respectively, confirmed after 3 months.
The core study consisted of Periods 1 and 2 where participants were scheduled for 12 intravenous (IV) infusions administered over 2 hours separated by 4-week intervals (Figure 1). In Period 1, participants received 6, 12, or 18 mg/kg temelimab, or placebo. In Period 2, participants in the placebo group were re-randomized in equal portions to one of the active treatment groups; the dosing scheme of Period 2 was continued during the extension study (Figure 1).
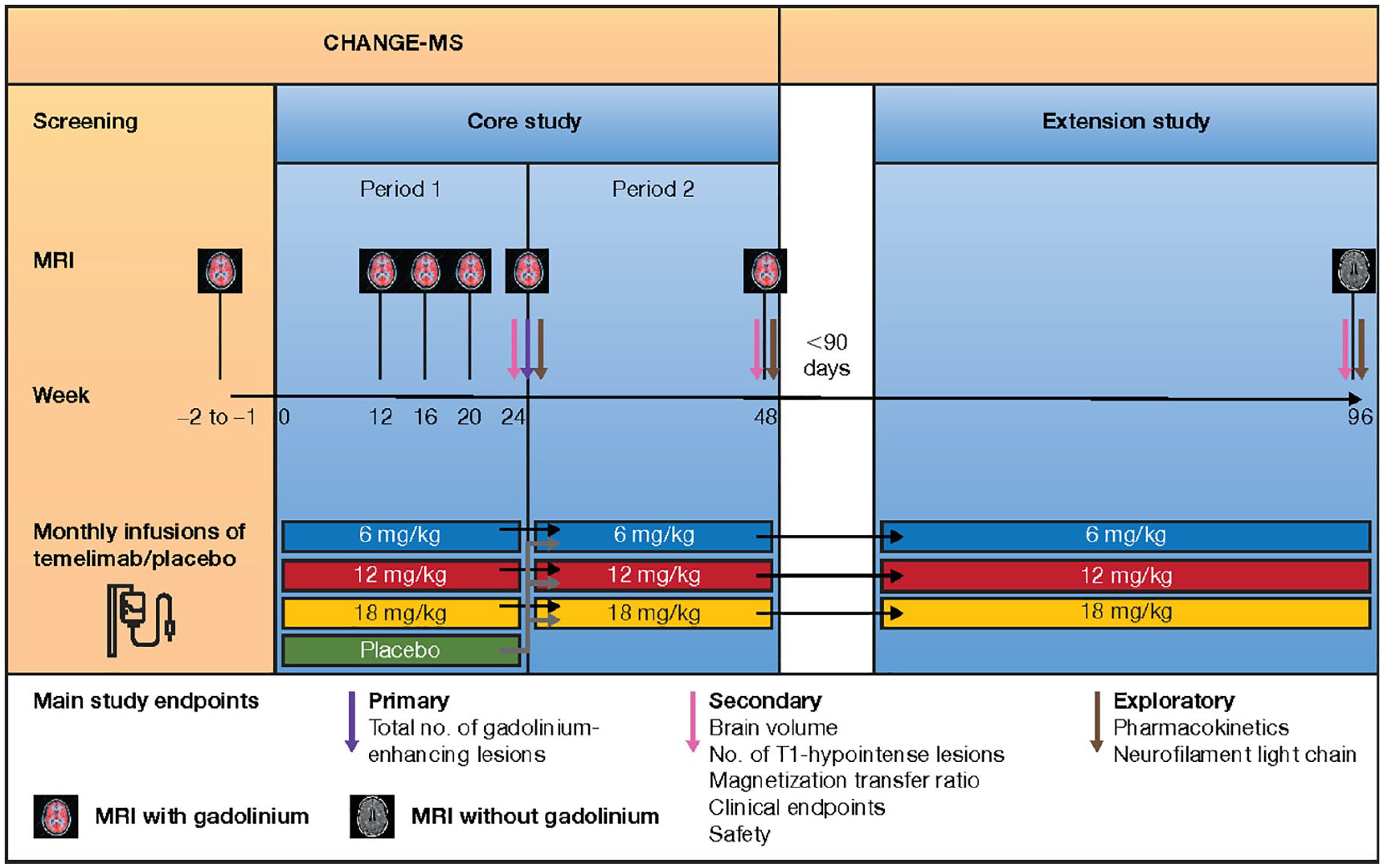
Study design. In Period 1, patients were randomly assigned in a 1:1:1:1 ratio to receive 1 of the 3 doses of temelimab or placebo for 24 weeks. In Period 2, participants from the placebo group of Period 1 were re-randomized to either of the 3 dose groups of temelimab in a 1:1:1 ratio, whereas participants in the temelimab dose groups continued to receive the same dose of temelimab. This dosing was continued in the extension study.
Treatment allocation was randomized centrally using an interactive web response system and blinded to participants, investigators, relevant site staff, magnetic resonance imaging (MRI) readers, and monitors for the duration, and to the sponsor until week 24.
Standard protocol approval, registrations, and participant consent
The study protocol was approved by local independent ethics committees. The study was conducted in accordance with Good Clinical Practice and the European Medicines Agency’s guideline on clinical investigation of medicinal products for the treatment of MS. Participants provided written informed consent before enrolment.
Endpoints
The primary endpoint, assessed at week 24 (end of Period 1), was the cumulative number of GdE T1 lesions identified on 4 monthly MRI scans from week 12 to 24. Secondary endpoints are presented in Table 1. 15
Drug safety and adverse events (AEs) were assessed at each visit.
Statistical analysis
Sample size and power calculations were done for the primary endpoint of GdE T1 lesions from week 12 to 24 using a simulation-based method based on a negative binomial generalized linear model (NB-GLM) with the following assumptions: expected cumulative mean number of lesions in placebo group: 4; common dispersion parameter (shape): 0.5; expected cumulative mean number of lesions in temelimab 18 mg/kg group: 1.6 (expected treatment effect: 60%, based on other RRMS drug trials). With 61 evaluable participants per treatment group, the expected power using a 2.5% one-sided type I error rate was 90%. Assuming a 5% attrition rate, 65 participants per group, or 260 overall, were required.
Each temelimab dose group was compared sequentially with placebo in the per protocol set-like population (participants with assessable MRIs at baseline and week 24, not missing ⩾2 consecutive MRIs between weeks 12 and 24) on the sum of new GdE T1 lesions from 4 MRIs from weeks 12 to 24, adjusted for the binary covariate absence or presence of GdE T1 lesions at baseline. Ratios of counts were calculated for each temelimab dose versus placebo. Overall, the one-sided type I error rate was controlled at 2.5% according to Wiens 16 method with the following fixed test sequence of temelimab versus placebo: 18 mg/kg, 12 mg/kg, then 6 mg/kg, until a null hypothesis could not be rejected.
Secondary analyses of T1 and T2 lesions were performed using an NB-GLM with adjustment for absence or presence of GdE T1 lesions at baseline. Ratios of counts were calculated for each temelimab dose versus placebo. Trend analyses by dose for the brain volume changes, and magnetization transfer ratio (MTR) at weeks 24, 48, and 96 were calculated by linear regression with a constant and 1 factor (coded 0 to 3) representing the different temelimab doses; least squares mean differences were calculated using this model, median percent changes were shown graphically. Secondary efficacy outcomes at week 48 were prespecified in the statistical analysis plan prior to database lock. Six potential exploratory analyses were defined prior to the week 48 database lock to compare different dosing regimens. Of these, it was decided, post hoc, to compare efficacy outcomes at weeks 48 and 96 in participants who received temelimab (any dose) in Periods 1 and 2 (“continuous temelimab”) with those in participants who received placebo in Period 1 followed by any dose of temelimab in Period 2 (the “placebo/comparator” group). In a further post hoc analysis, the subgroup of participants with inactive disease (no GdE T1 lesions ⩾ 3 mm at baseline) was analysed. In the extension study, participants who terminated the study before or after week 96 had an end of study visit, and their data were carried forward or backward to 96 weeks. Except for the primary endpoint, all analyses were two-sided with an alpha error of 5%. Corrections for multiple comparisons were not performed on secondary or exploratory analyses. SAS version 9.3 software was used.
Results
Disposition and baseline characteristics
The disposition of study participants is shown in Figure 2. Overall, 270 participants were randomized to receive monthly IV temelimab (6, 12, or 18 mg/kg), or placebo; 2 participants did not receive study drug (1 each in the placebo and 12 mg/kg temelimab groups). The full analysis set comprised 263 participants as 7 did not complete any post-baseline assessments (2 each in the 12 mg/kg and 18 mg/kg temelimab groups, and 3 in the placebo group). In total, 247 participants completed Period 1 (Figure 2(a)) and continued to Period 2. Of 232 participants who completed the “core study” (Periods 1 and 2) (Figure 2(b)), 220 entered the extension study (1 did not receive study drug), and 154 completed ⩾48 weeks of the extension (⩾96weeks total). The remaining participants discontinued before 96 weeks due to the early termination of the study (Figure 2(c)).
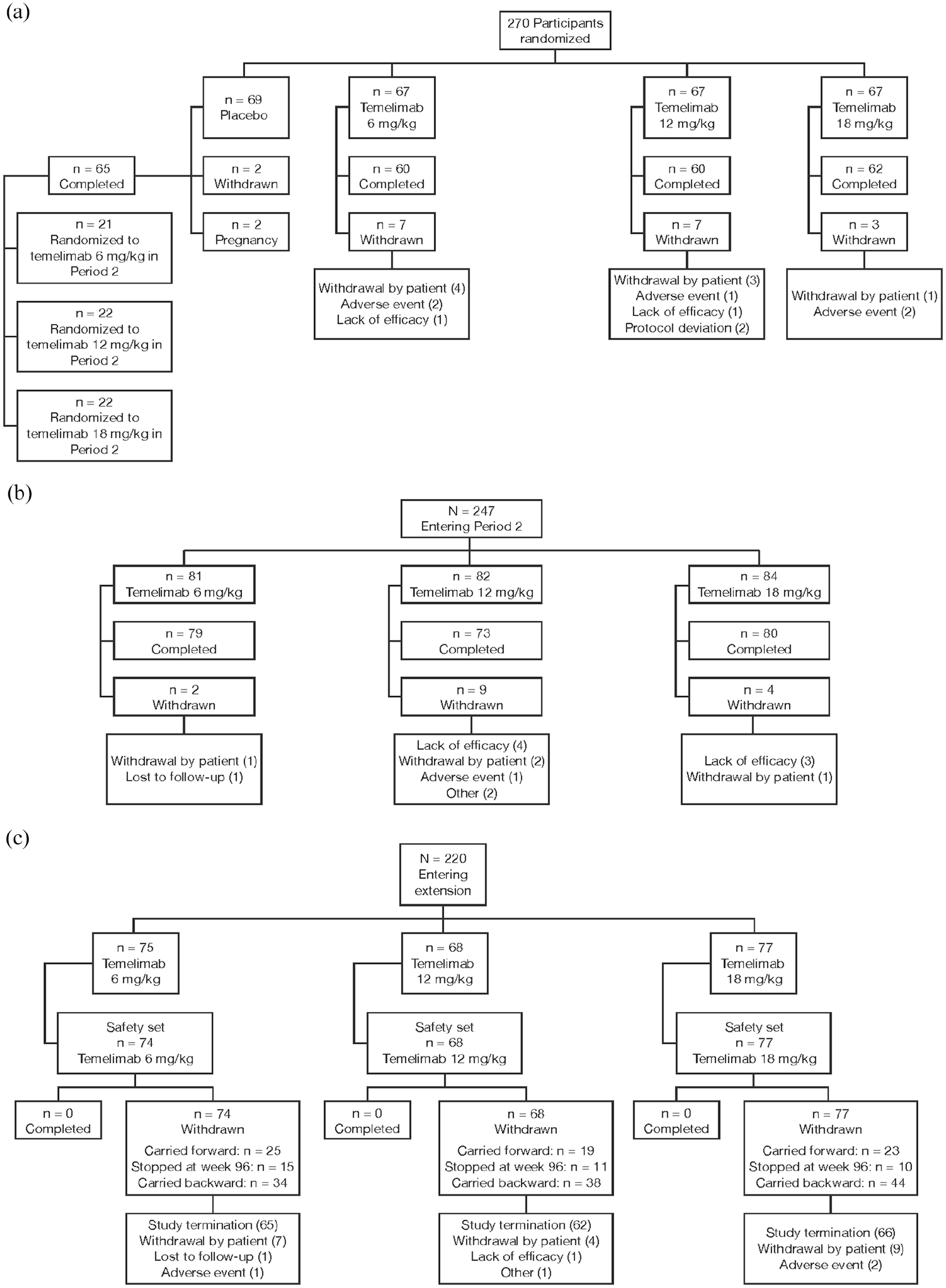
Patient disposition: core study Periods 1 (a) and 2 (b), and extension study (c).
Participant demographics and disease characteristics at baseline were similar across the treatment groups, apart from the placebo group which had a shorter disease duration compared with the three active treatment groups (p = 0.012); however, the difference between the placebo group and the 18 mg/kg group was not statistically significant (Table 2). Gadolinium-enhancing lesions at baseline were more numerous in the placebo group, but this was not significant either for each active treatment arm versus placebo, nor for the comparison of all active treatment arms versus placebo (p = 0.915) (Table 2).
Participant characteristics at baseline (core study).
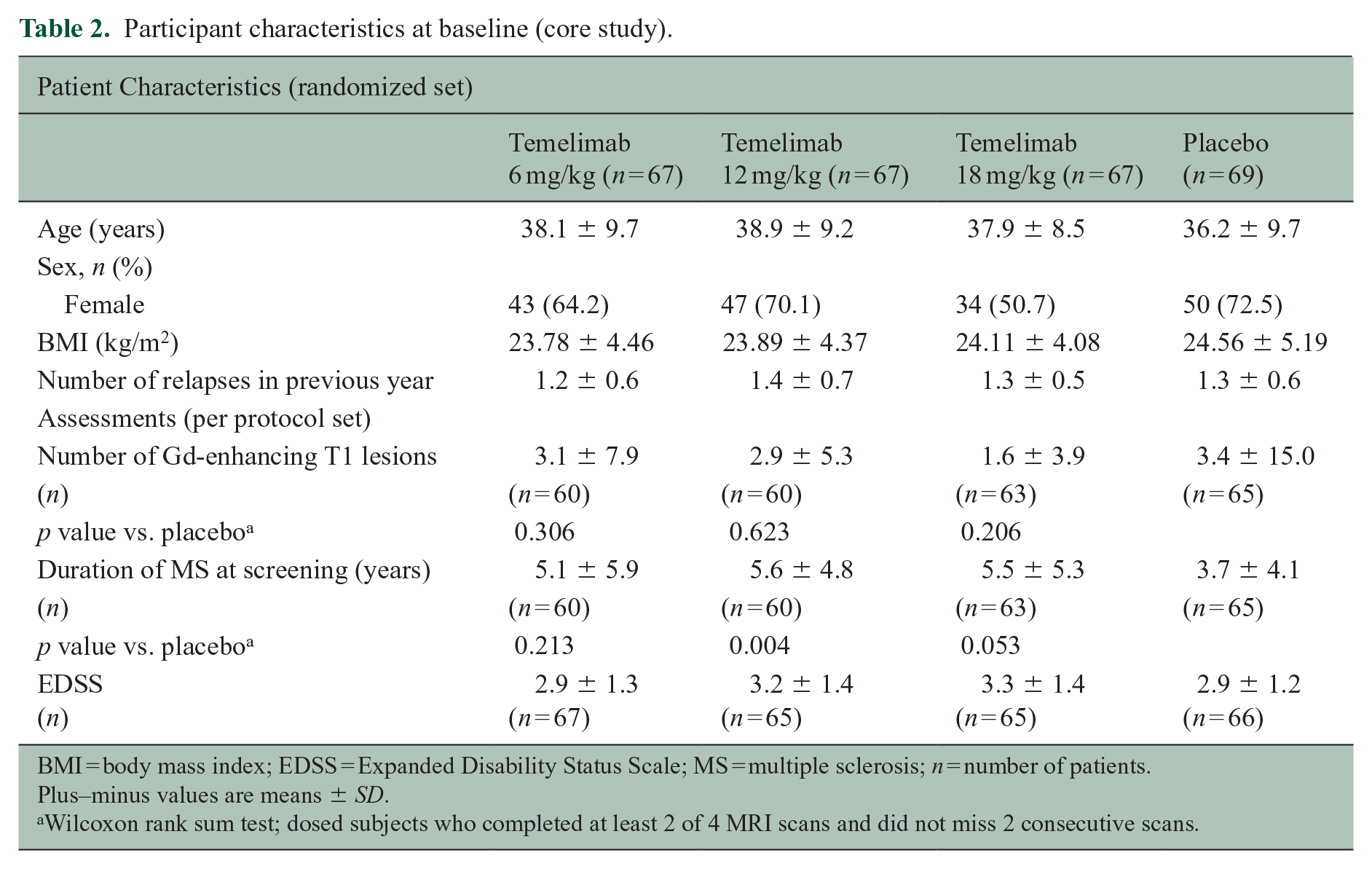
BMI = body mass index; EDSS = Expanded Disability Status Scale; MS = multiple sclerosis; n = number of patients.
Plus–minus values are means ± SD.
Wilcoxon rank sum test; dosed subjects who completed at least 2 of 4 MRI scans and did not miss 2 consecutive scans.
Overall, 90% of participants had not received any disease modifying therapy (DMT) prior to the study.
Imaging
At week 24, there was no statistically significant difference between temelimab doses and placebo for the primary efficacy endpoint of cumulative number of GdE T1 lesions from week 12 to 24, after adjusting for presence/absence of baseline activity (Table 3). This was similar after adjusting for disease duration at screening (pairwise comparison of temelimab vs. placebo groups, data not shown). The proportion of participants free of GdE T1 lesions from week 12 to 48 was significantly larger in the continuous 18 mg/kg temelimab group (46.8%) versus the placebo/comparator group (all participants initially randomized to placebo and re-randomized to any dose of temelimab in Period 2; 27.7%) with a treatment difference of 0.19 (95% confidence interval [CI] 0.03–0.36; p = 0.029). There were no significant differences between treatment groups for the number of new and/or enlarging T2 lesions on brain MRI scans from weeks 12 to 24 (Period 1), weeks 24 to 48 (Period 2), or from weeks 48 to 96 of the extension study. While at baseline, the continuous 18 mg/kg temelimab and the placebo/comparator groups had similar numbers of qualifying T1-hypointense lesions, at week 48 significantly fewer new lesions occurred in the continuous 18 mg/kg temelimab group (Table 4). No significant effect for this measure was seen with either 6 or 12 mg/kg continuous temelimab versus the placebo/comparator group.
Cumulative number of gadolinium-enhancing T1 lesions from week 12 to 24 (per protocol set-like population).
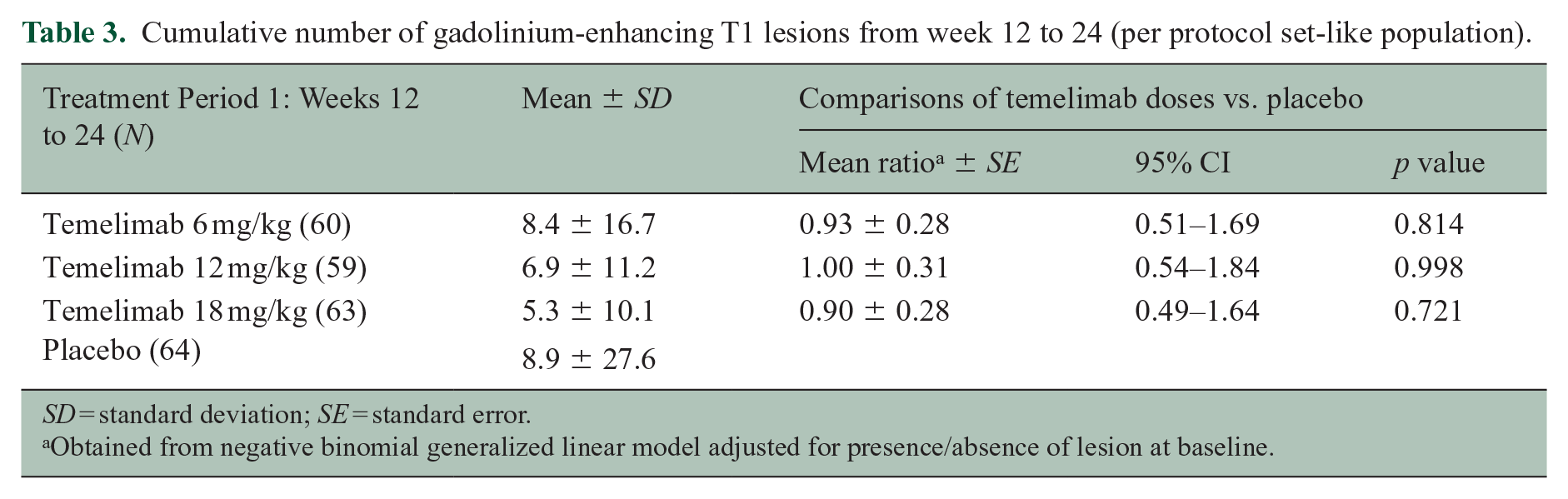
SD = standard deviation; SE = standard error.
Obtained from negative binomial generalized linear model adjusted for presence/absence of lesion at baseline.
Number of qualifying new T1 hypointense lesions from baseline to week 48 (full analysis set entering Period 2).
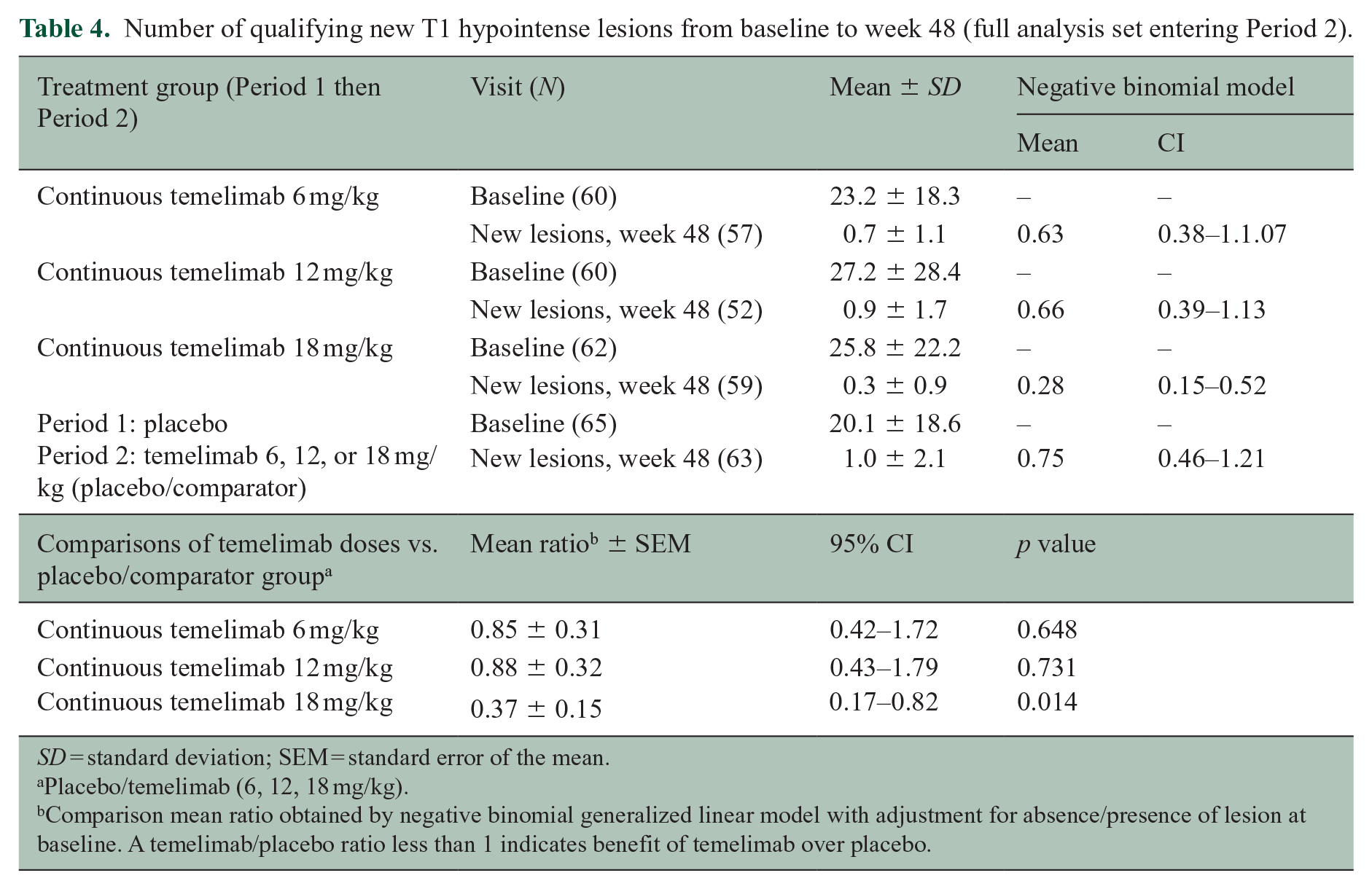
SD = standard deviation; SEM = standard error of the mean.
Placebo/temelimab (6, 12, 18 mg/kg).
Comparison mean ratio obtained by negative binomial generalized linear model with adjustment for absence/presence of lesion at baseline. A temelimab/placebo ratio less than 1 indicates benefit of temelimab over placebo.
The median whole brain volume loss in participants treated with continuous 18 mg/kg temelimab versus the placebo/comparator group was reduced by 27.1% at week 48 (core study) and 15.4% at week 96 (extension study) (Figure 3(a)). Accordingly, the median volume loss of cerebral cortex and thalamus was reduced by 31.3% and 71.6% at week 48, and 41.9% and 42.6% at week 96, respectively (Figure 3(b) and (c)). In a post hoc analysis, we observed a dose-response effect for continuous temelimab versus the placebo/comparator group for thalamic volume at week 48 (p = 0.01) and week 96 (p = 0.04), and a dose-response trend for cortical volume (p = 0.10; p = 0.06, respectively) in these periods, but not for whole brain volume (p = 0.26; p = 0.46, respectively). Furthermore, analysis of core study data restricted to the subgroup of participants with inactive disease (without T1 GdE lesions at baseline) showed similar effects on atrophy in the temelimab 18 mg/kg group versus placebo/comparator over 48 weeks. Whole brain, cortical, and thalamic volume loss was reduced by 38%, 49%, and 72% versus placebo/comparator (Spearman rank correlation p = 0.132, p = 0.025, p = 0.575), respectively. Participants treated with continuous 18 mg/kg temelimab had a minimal decrease or stabilization of the MTR signal across prespecified bands in the periventricular normal-appearing white matter (NAWM), cortex, and whole brain at weeks 24, 48, and 96, compared with respective declining signal changes in the placebo/comparator group, as measured by linear regression (Table 5). For 10 of 14 MTR measures, this was statistically significant at the 48- and 96-week time points.
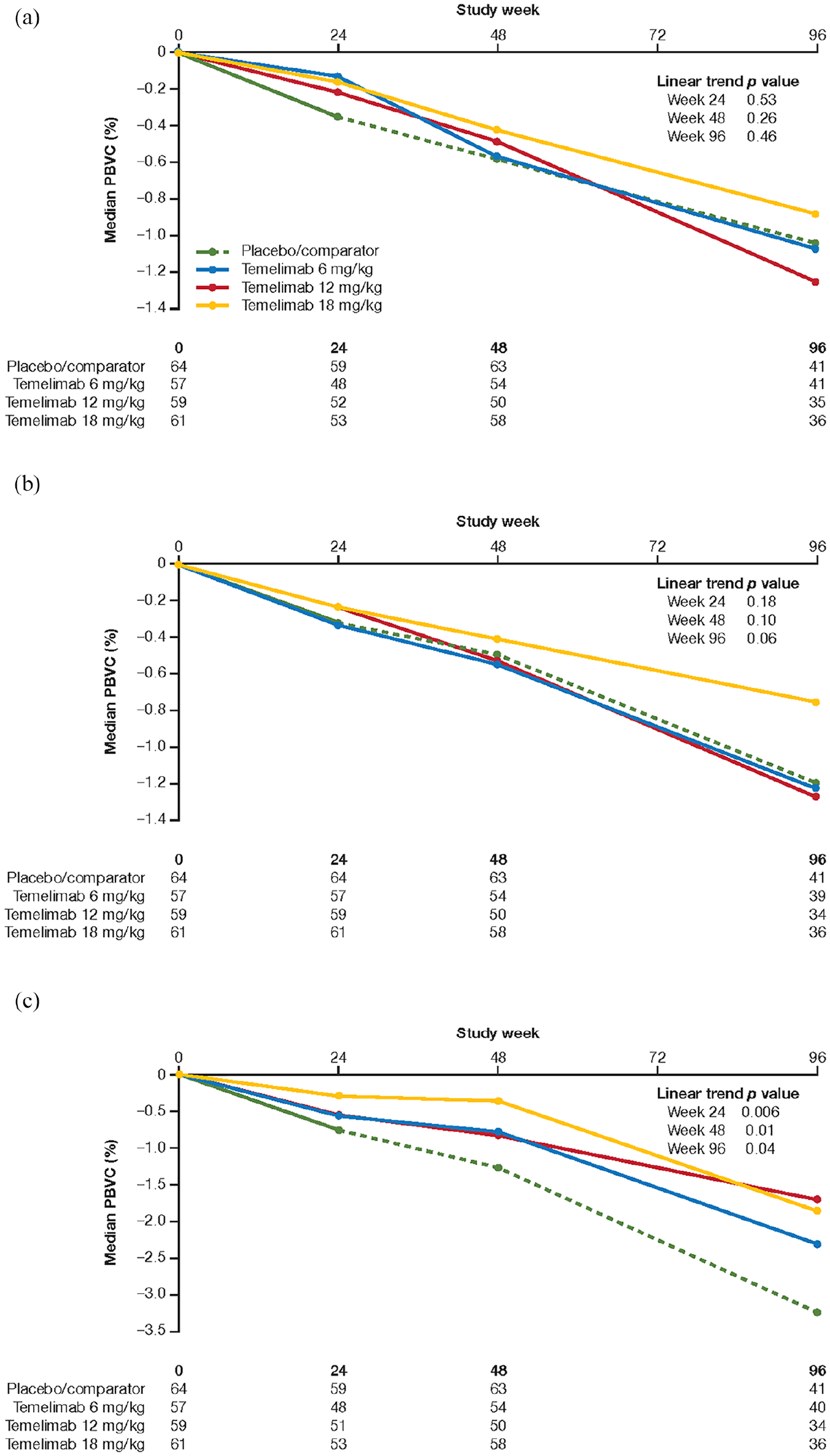
Percentage change from baseline in (a) whole brain, (b) cortical, and (c) thalamic volume. Percentage change from baseline to week 48 of the core study and to week 96/end of extension study (full analysis set entering Period 2). Linear trends were analysed with a linear regression model including an intercept and a parameter for doses. Dashed line indicates period after rerandomization of patients originally randomized to placebo to 1 of the three temelimab doses. The number of patients available for assessment at each time point is presented by treatment group below each figure.
Magnetization transfer ratio in periventricular bands, cerebral cortex bands, and whole brain at weeks 24, 48, and 96.
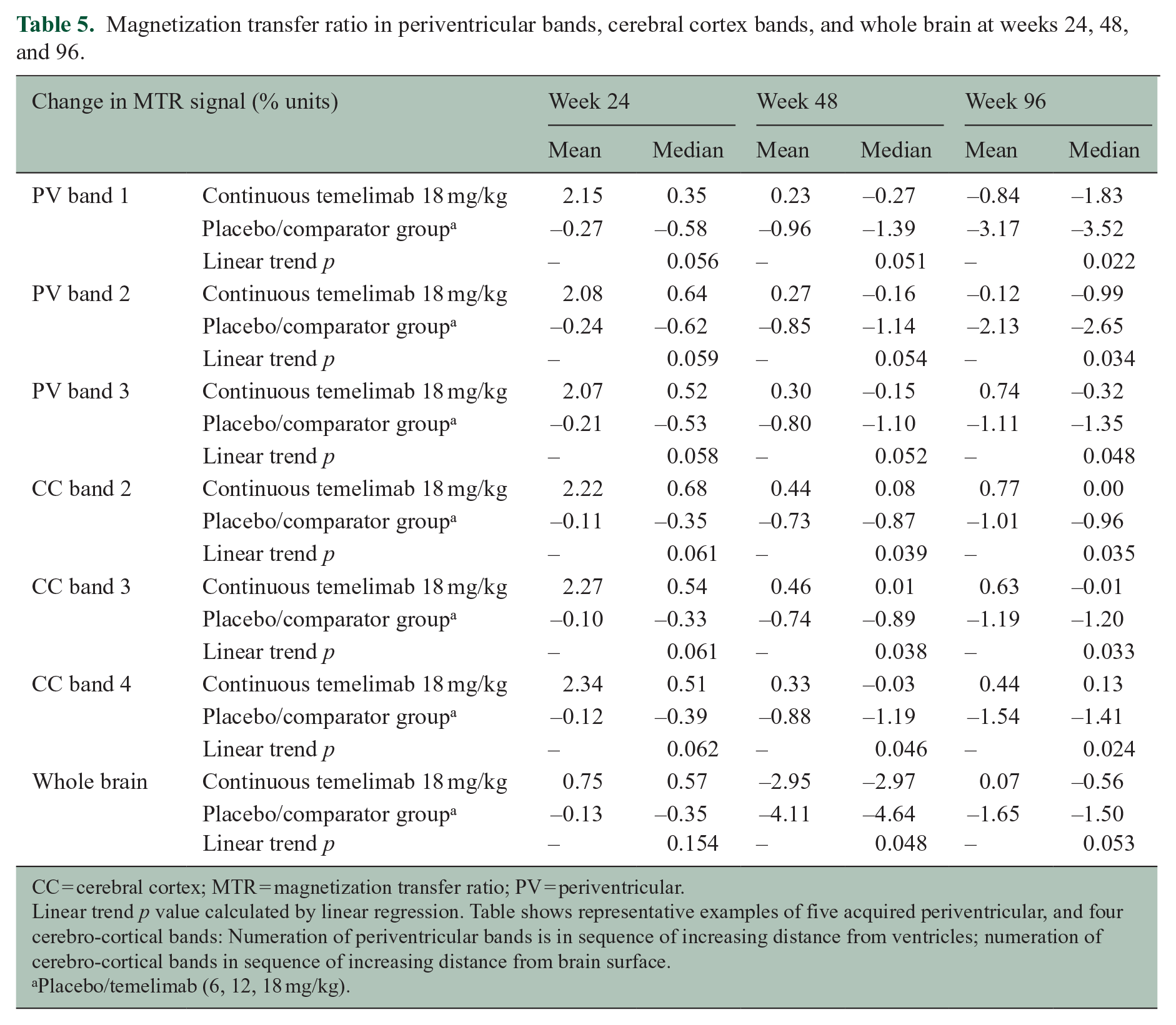
CC = cerebral cortex; MTR = magnetization transfer ratio; PV = periventricular.
Linear trend p value calculated by linear regression. Table shows representative examples of five acquired periventricular, and four cerebro-cortical bands: Numeration of periventricular bands is in sequence of increasing distance from ventricles; numeration of cerebro-cortical bands in sequence of increasing distance from brain surface.
Placebo/temelimab (6, 12, 18 mg/kg).
Clinical measures
There were no significant differences between treatment groups regarding annualized relapse rate (ARR) in the core study (at week 48 (range 0.60–0.66)) or the extension (at week 96 (range 0.71–0.78)), or for the proportion of participants free of relapse in all treatment groups. Disability progression, as measured by change in EDSS from baseline to week 96, was not different across groups with 8.3% (4/48), 4.8% (2/42), 3.8% (2/53), and 9.1% (5/55) participants, for 6, 12, and 18 mg/kg temelimab, and placebo/comparator groups, respectively. There was no difference in confirmed disability improvement and number of participants with no evidence of disease activity (NEDA) across treatment groups.
Safety
There were no significant differences in treatment-emergent AEs between treatment groups or when temelimab treatment groups were compared with the placebo group in Period 1. Twenty-two participants experienced 26 serious AEs (SAEs) (Supplementary Table 2) during the core study and the extension study; 10 SAEs occurred during the core study, of which one was considered as “related to treatment” (one participant treated with temelimab 12 mg/kg experienced macroscopic hematuria). Two SAEs occurred in participants treated with placebo: one participant with a cholecystitis and one with a rib fracture. The most commonly reported treatment-emergent AEs concerned the System Organ Class (SOC) “Infections and Infestations.” In the placebo-controlled Period 1, 19.4%, 19.7%, 22.4%, and 22.1% of participants treated with 6 mg/kg, 12 mg/kg, 18 mg/kg temelimab, and placebo, respectively, presented with an infection (the most frequent were nasopharyngitis and upper respiratory tract infections). During the entire core study, 27.3%, 31.1%, and 32.6% of participants in the temelimab 6 mg/kg, 12 mg/kg, and 18 mg/kg groups, respectively, presented with an infection.
No differences were observed between treatment groups for infusion-related reactions during the core study. These reactions were all mild or moderate in severity.
There were no differences between treatment groups in results of laboratory evaluations, vital signs (including cardiac assessments), physical examinations, or Columbia-Suicide Severity Rating Scale.
During the extension study, 16 SAEs occurred in 12 participants, of which two were considered treatment-related (breast cancer and toxic hepatitis in one participant each, treated with 18 mg/kg temelimab). The time between start of treatment with temelimab and the breast cancer diagnosis was 20 months. The participant with toxic hepatitis showed initial improvement following the end of temelimab administration; however, the symptomatology recurred 3.5 months later. One death occurred in a participant presenting with two SAEs (bilateral pneumonia and endotoxemia, considered unrelated to treatment by the investigator). Except for the information on the death certificate, all information received was reported by the participant’s relatives. As no further medical information could be obtained, the sponsor upgraded these events to suspected unexpected serious adverse reactions thereby allowing expedited reporting. The most frequently reported AEs were in the SOC “Infections and Infestations” (mainly nasopharyngitis and upper respiratory tract infections), occurring in 21.6%, 25.0%, and 32.5% of participants in 6 mg/kg, 12 mg/kg and 18 mg/kg temelimab groups, respectively. The second most frequent AEs were in the SOC “Musculoskeletal and Connective Tissue disorders,” with 9.5%, 7.4%, and 6.5% of participants in the 6 mg/kg, 12 mg/kg, and 18 mg/kg temelimab groups, respectively. Only one potential infusion-related reaction was reported; hypotension starting several hours after the end of the infusion. No meaningful differences in laboratory evaluations, vital signs, physical examinations, EKG, or C-SSRS scores between treatment groups emerged during the extension. Antibodies against temelimab were detected in two participants: one became positive and then reverted to negative status and one became positive at the termination visit.
Discussion
The primary endpoint of the core study, cumulative number of GdE T1 lesions at 24 weeks, was not met. This may indicate that temelimab, at the doses tested, had no or only marginal effect on this measure of neuroinflammation. However, the results provide preliminary MRI evidence that temelimab could exert an anti-neurodegenerative effect based on measures of brain atrophy, myelin integrity, and number of chronic T1 hypointense lesions.
Preclinical studies suggest that HERV-W-ENV exerts pro-inflammatory effects in monocytes and dendritic cells via binding to the Toll-like receptor-4/cluster of differentiation 14 receptor complex, which could be antagonized by a neutralizing antibody.7,8 The hypothesis of a pathogenic role of HERV-W-ENV in MS was based on three observations: (a) its marked expression in brain tissue of MS patients,4–6 (b) results of experimental autoimmune encephalomyelitis (EAE) studies where HERV-W-ENV worsened myelin oligodendrocyte glycoprotein–EAE, which was ameliorated by temelimab, 17 and (c) in vitro models that demonstrated toxic and dystrophic effects on neurons and OPCs,4,5 respectively, which were also antagonized by temelimab.7,9,11 Based on these observations, this study used MRI-based measures of inflammation at 24 weeks (i.e. GdE T1 and T2 lesions), as a typical primary endpoint for phase 2 studies of immunomodulatory agents in relapsing MS, 12 and MRI measures of neurodegeneration (brain volume, MTR, T1-hypointense lesions) at 48 and 96 weeks as secondary endpoints. An important learning from the present study is that hypothesized modes of action based on preclinical models, 17 here inhibition of immune activation by HERV-W-ENV by temelimab, may not translate into clinical effects on acute inflammation in MS.
As an alternative mode of action, the results suggest that neutralization of HERV-W-ENV might have a neuroprotective effect in patients with RRMS. Results of the core study (reduction of brain atrophy, reduced formation of T1-hypointense lesions, and stabilization of MTR signal) suggest a possible, dose-dependent radiographic effect of temelimab at the highest dose of 18 mg/kg, compared with the placebo/comparator group. These findings became more evident during the extension study at 96 weeks. The fact that the subgroup of inactive (no GdE lesions at baseline) participants treated with 18 mg/kg temelimab showed a similar relative reduction in whole and regional brain atrophy as the overall study group could be interpreted as supportive evidence of a neuroprotective capacity independent of an effect on inflammation caused by the adaptive immune system. HERV-W-ENV is expressed in active and chronic MS lesions 6 ; we hypothesize that clinically effective target engagement of temelimab occurs there, attenuating the neurodegenerative effect of these lesions. The study was not powered for these secondary and exploratory endpoints and while many of the MRI outcomes were not, or only marginally, statistically significant, they were consistent.
Participants in the initial placebo arm had shorter disease duration and more Gd-enhancing lesions at baseline than those in the temelimab arms. Both features are indicative of higher disease activity and this constellation could potentially lead to a reduced effect size for anti-inflammatory DMT versus placebo. However, these imbalances were not significant for the comparison of the 18 mg/kg treatment arm versus placebo. Considering that temelimab did not show anti-inflammatory properties, we conclude that these differences did not impact on the results.
Previous studies have found that where more GdE T1 lesions are present, there is a stronger “pseudoatrophy effect” with an anti-inflammatory DMT, which could confound whole brain volume analysis. 18 Based on our current understanding of temelimab’s mechanism of action (i.e. that it has no anti-inflammatory effect), results are not likely to be biased by pseudoatrophy. This is also evidenced by a similar brain volume loss as with placebo in the first 24 weeks (when pseudoatrophy is typically most pronounced 18 ), a similar pattern of atrophy in grey versus white matter, 19 and a similar course of atrophy in the subgroup of inactive patients.
Repeated administrations of temelimab were well tolerated. Very few SAEs occurred with no differences in the type or frequency of AEs emerging between treatment groups. Infusion-related reactions were mild or moderate, supporting the planned long-term administration of temelimab needed to treat MS and the potential for its use in combination with other DMTs.
This study has several limitations. The study population had relevant inflammatory disease activity before and during the trial that may have masked potential neuroprotective effects of temelimab. A conservative maximum dose of 18 mg/kg was used in this study. Notably, effects were primarily observed at this dose and further optimization is therefore needed to define the minimal maximally effective dose. The re-randomization of participants from placebo to active treatment after week 24 and a sample size with limited statistical power for exploratory MRI outcomes may have attenuated the effect size of temelimab treatment. The lack of correction for multiple comparisons across secondary endpoints suggests the possibility for type I errors.
In summary, temelimab failed to show a significant effect on the cumulative number of T1 GdE lesions at the doses studied. However, preliminary evidence indicates that neutralization of HERV-W-ENV protein could have positive effects on MRI measures of neurodegeneration in MS.
Two primary limitations of the current study (the need for dose optimization and interference of preexisting/comorbid inflammatory disease activity) will be addressed in a new phase 2 study, with an extended dose range of temelimab in MS patients whose acute disease activity has been reset by chronic anti-CD20-Ab therapy, but who still experience clinical progression (ClinicalTrials.gov: NCT04480307).
Supplemental Material
sj-pdf-1-msj-10.1177_13524585211024997 – Supplemental material for Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study
Supplemental material, sj-pdf-1-msj-10.1177_13524585211024997 for Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study by Hans-Peter Hartung, Tobias Derfuss, Bruce AC Cree, Maria Pia Sormani, Krzysztof Selmaj, Jonathan Stutters, Ferran Prados, David MacManus, Hans-Martin Schneble, Estelle Lambert, Hervé Porchet, Robert Glanzman, David Warne, Francois Curtin, Gabrielle Kornmann, Bénédicte Buffet, David Kremer, Patrick Küry, David Leppert, Thomas Rückle and Frederik Barkhof in Multiple Sclerosis Journal
Supplemental Material
sj-pdf-2-msj-10.1177_13524585211024997 – Supplemental material for Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study
Supplemental material, sj-pdf-2-msj-10.1177_13524585211024997 for Efficacy and safety of temelimab in multiple sclerosis: Results of a randomized phase 2b and extension study by Hans-Peter Hartung, Tobias Derfuss, Bruce AC Cree, Maria Pia Sormani, Krzysztof Selmaj, Jonathan Stutters, Ferran Prados, David MacManus, Hans-Martin Schneble, Estelle Lambert, Hervé Porchet, Robert Glanzman, David Warne, Francois Curtin, Gabrielle Kornmann, Bénédicte Buffet, David Kremer, Patrick Küry, David Leppert, Thomas Rückle and Frederik Barkhof in Multiple Sclerosis Journal
Footnotes
Acknowledgements
Estelle Lambert who conducted the statistical analysis of the results of the CHANGE-MS study and reviewed a previous version of the manuscript reporting data from that study only, is employed by Servier Laboratories (Neuilly-sur-Seine, France). GeNeuro SA funded Totzke & Dreher Scientfic SA (Geneva, Switzerland) (CHANGE-MS data) and Andrea Plant, PhD, of Caudex (Oxford, UK) (CHANGE-MS plus ANGEL-MS data) to assist with finalizing the manuscript under author instruction from material prepared by Dr. Glanzman and Dr. Curtin, with review and editorial comments from all authors during the process. Editorial support, funded by GeNeuro, was provided by Jonathan Robertson of Scinopsis Ltd. (Brighton, UK). Dr. Barkhof is supported by the National Institute for Health Research University College London Hospitals NHS Foundation Trust Biomedical Research Centre. Dr. Prados held a Guarantors of Brain non-clinical postdoctoral fellowship.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The study was sponsored by GeNeuro SA (Geneva, Switzerland). H.-P. Hartung has received honoraria for consulting and serving on steering and data monitoring committees from Bayer Healthcare, Biogen, Celgene Receptos BMS, GeNeuro SA, MedDay, MedImmune, Merck, Novartis, Roche, Sanofi Genzyme, Teva, TG Therapeutics, and VielaBio with permission by the rector of Heinrich-Heine-Universität Düsseldorf. T. Derfuss has received grants from Biogen and Novartis, and his institution receives financial compensation for activities as a steering committee member from Actelion, Alexion, Biogen, Celgene, GeNeuro SA, MedDay, Merck, Mitsubishi Pharma, Novartis, Roche, and Sanofi Genzyme. B.A.C. Cree has received personal compensation for consulting from Alexion, Atara, Biogen, EMD Serono, Novartis, Sanofi, and TG Therapeutics. M.P. Sormani has received personal compensation for consulting from Biogen, Celgene, GeNeuro SA, Immunic, MedDay, Merck, Mylan, Novartis, Roche, Sanofi, and TEVA. K. Selmaj has received honoraria for speaking, consulting and serving on advisory boards for Biogen, Celgene, Genzyme, Merck, Mylan, Novartis, Roche, and TG Therapeutics. J. Stutters received grants from BioClinica, Inc., during the conduct of the study. F. Prados has received a personal fellowship grant from the non-clinical Guarantors of Brain and personal fees from BioClinica, Inc. D. MacManus received grants from BioClinica, Inc., during the conduct of the study. H.-M. Schneble was an employee of Servier, the study sponsor’s development partner for these studies and is currently an employee of Roche. E. Lambert is an employee of Servier, the study sponsor’s development partner for these studies. H. Porchet, R. Glanzman, F. Curtin, and G. Kornmann were employees of the sponsor, GeNeuro SA, during the execution of the study. D. Warne has received personal fees for statistical work on the study. P. Küry performed consultancy work for GeNeuro SA and is supported by the Stifterverband/Novartisstiftung. F. Barkhof has received grants and other from Apitope, Biogen, GE Healthcare, GeNeuro SA, IXICO, Merck, Novartis, Roche, and Teva. G. Francis, B. Buffet, D. Leppert, and T. Rückle are employees of the study sponsor, GeNeuro SA.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Both studies were funded by GeNeuro SA and Servier. The sponsor and the principal investigator (H.-P. Hartung) designed the protocol and were responsible for the study conduct. The corresponding author had full access to all of the study data and had final responsibility for the decision to publish. Writing and editorial support was funded by GeNeuro SA. No honoraria, grants, or other forms of payment were provided to investigators, with the exception of fees paid in connection with the performance of the studies.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.