
Abstract
Background:
Progressive multifocal leukoencephalopathy (PML) can in rare cases occur in natalizumab-treated patients with high serum anti-JCPyV antibodies, hypothetically due to excessive blockade of immune cell migration.
Objective:
Immune cell recruitment to the central nervous system (CNS) was assessed in relapsing-remitting multiple sclerosis (RRMS) patients stratified by low versus high anti-JCPyV antibody titers as indicator for PML risk.
Methods:
Cerebrospinal fluid (CSF) cell counts of 145 RRMS patients were quantified by flow cytometry. Generalized linear models were employed to assess influence of age, sex, disease duration, Expanded Disability Status Scale (EDSS), clinical/radiological activity, current steroid or natalizumab treatment, as well as anti-JCPyV serology on CSF cell subset counts.
Results:
While clinical/radiological activity was associated with increased CD4, natural killer (NK), B and plasma cell counts, natalizumab therapy reduced all subpopulations except monocytes. With and without natalizumab therapy, patients with high anti-JCPyV serum titers presented with increased CSF T-cell counts compared to patients with low anti-JCPyV serum titers. In contrast, PML patients assessed before (n = 2) or at diagnosis (n = 5) presented with comparably low CD8 and B-cell counts, which increased after plasma exchange (n = 4).
Conclusion:
High anti-JCPyV indices, which could be indicative of increased viral activity, are associated with elevated immune cell recruitment to the CNS. Its excessive impairment in conjunction with viral activity could predispose for PML development.
Keywords
Introduction
Natalizumab treatment can in rare cases be associated with the development of progressive multifocal leukoencephalopathy (PML). Current PML risk stratification includes the factors long-term treatment with natalizumab, 1 prior immune suppression and the presence of anti-JCPyV antibodies in serum. 2 Natalizumab interferes with lymphocyte trafficking into the gut 3 and central nervous system (CNS). 4 Therapy is associated with an elevated anti-JCPyV antibody seroconversion rate, 5 indicating that natalizumab might modulate JCPyV immune control in peripheral tissues such as the gut, which has been shown to be a primary reservoir for JCPyV. 6 Other previously proposed, independent PML biomarkers point towards a similar hypothesis: a lack of intrathecal, lipid-specific IgM bands and lack of L-selectin on peripheral CD4 T cells might both indicate suboptimal localized immune control due to insufficient peripheral immune cell recruitment.7,8 Current risk mitigation strategies include extending the natalizumab dosing intervals, hypothetically to allow for a window of localized immune cell recruitment and surveillance. 9 Practically all natalizumab-associated PML cases so far have been JCPyV-seropositive, 87% with ‘JCV index’ values (normalized anti-JCPyV antibody titre) above 1.5 more than 6 months before PML diagnosis. 2 This suggests increased viral activity in JCPyV-seropositive patients, which might allow for development of viral mutations necessary for pathogenicity. 10 However, as only a minor fraction of high-risk patients (‘JCV index’ > 1.5) eventually develops PML, it is conceivable that high viral activity needs to coincide with continuously insufficient immune cell recruitment to affected tissue, finally resulting in breakdown of tissue-restricted, JCPyV-specific immune control in later PML patients. The concept of sufficient peripheral immune cell recruitment being a prerequisite for maintaining localized CD8-mediated immune control in the CNS 11 is supported by a recent murine study, showing that continuous infiltration of CD4 T cells is necessary to prevent CNS polyomavirus pathology resembling PML. 12
The goal of this study was, therefore, to assess CNS immune cell recruitment in the context of PML risk and to evaluate, whether (1) increased viral activity/suboptimal viral control (as indicated by high ‘JCV index’ values) might be associated with elevated CNS immune cell abundance (as quantified by cerebrospinal fluid (CSF) leukocyte numbers) to sustain a localized immune defence and (2) PML development might be associated with abnormal CNS immune cell recruitment.7,13
Methods
145 MS patients, who either did not receive immune modulatory therapy at that time (at least 2 months before lumbar puncture) or were under long-term (>18 months) therapy with natalizumab, and five patients developing PML alongside natalizumab therapy, were included in the study between 2010 and 2019 (Table 1). All subjects underwent routine flow cytometry (FC) assessment of CSF immune cell subsets, and JCPyV serology was determined according to the Stratify-JCV assay (Unilabs, Copenhagen). 2 FC CSF analysis entailed antibodies against CD3, CD4, CD8, CD14, CD19, CD45, CD56, and CD138, allowing identification of the following leukocyte (CD45+) subpopulations: CD4 or CD8T cells (SSClow CD14– CD3+ CD56–, either CD4+ CD8– or CD4– CD8+), natural killer (NK) cells (SSClow CD14–CD3–CD56+), B-cells (SSClow CD14– CD19high CD138–), plasma cells (SSClow CD14– CD19low CD138high), and monocytes (SSCint CD14+). CSF cell counts for each cell type were calculated by flow count fluorospheres (Beckman Coulter) used as internal standard in each sample and subjected as dependents to generalized linear models (GLMs) with gamma-distributed errors and a log-link function. In the case of plasma cells as the dependent variable, a negative binomial model was used to account for the high number of samples with zero counts (55 samples (36%) contained 0 plasma cells per mL). Evaluated factors for the CSF cell count were sex, age, disease duration, Expanded Disability Status Scale (EDSS), current clinical activity (defined as a relapse within 6 weeks of lumbar puncture and/or radiological disease activity defined as either a contrast-enhancing lesion or a new T2-weighted lesion within 12 weeks of lumbar puncture), treatment with natalizumab, and JC virus (JCV) serology (binary; ‘JCV index’ <> 1.5 according to PML risk). The GLM provided two estimates: a p value to indicate, whether explanatory variables significantly influenced the dependent variable (e.g. CD4 T cell counts), which was false discovery rate (FDR)-corrected using two-stage step-up 1% FDR by Benjamini et al.; 14 and a coefficient, which is presented as an odds ratio (OR = ecoefficient of the main effect) for the binary factors as main effects. From six relapsing-remitting multiple sclerosis (RRMS) patients, two time points matched the inclusion criteria resulting in 151 modelled samples. Autocorrelation within subjects was modelled using an autoregressive (AR(1)) covariance matrix. Longitudinal data of five additional patients with natalizumab-associated PML were excluded from modelling. From all five patients, the sample at the time point of PML diagnosis (acute) was available; from two of these patients also a sample six or 26 months before PML diagnosis (pre), and from four patients a sample 3–7 weeks after diagnosis, directly after plasma exchange (post) was available.
Patient characteristics.
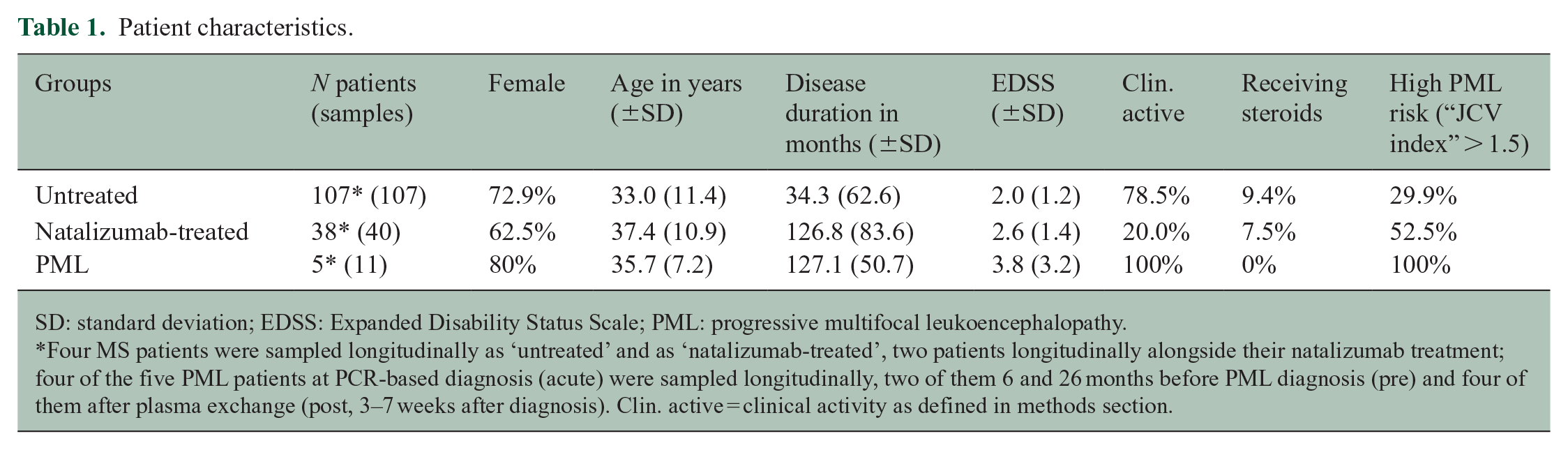
SD: standard deviation; EDSS: Expanded Disability Status Scale; PML: progressive multifocal leukoencephalopathy.
Four MS patients were sampled longitudinally as ‘untreated’ and as ‘natalizumab-treated’, two patients longitudinally alongside their natalizumab treatment; four of the five PML patients at PCR-based diagnosis (acute) were sampled longitudinally, two of them 6 and 26 months before PML diagnosis (pre) and four of them after plasma exchange (post, 3–7 weeks after diagnosis). Clin. active = clinical activity as defined in methods section.
To assess reliability of CSF cell count measurements via FC, they were correlated with routine microscopic cell counting using the Fuchs–Rosenthal chamber. There was strong correlation between the counting methods (Pearson rho 0.8131; Figure 1(a)). However, low cell count samples are often reported as 0 leukocytes/µL, which was the case in 29 samples of our study. The fact that FC could detect a median of 542.9 leukocytes/mL (0.54 leukocytes/µL) in these samples shows that ‘0 leukocytes/µL’ does not indicate an exact value of 0 but a value below the threshold of detection with sampled microscopic counting.
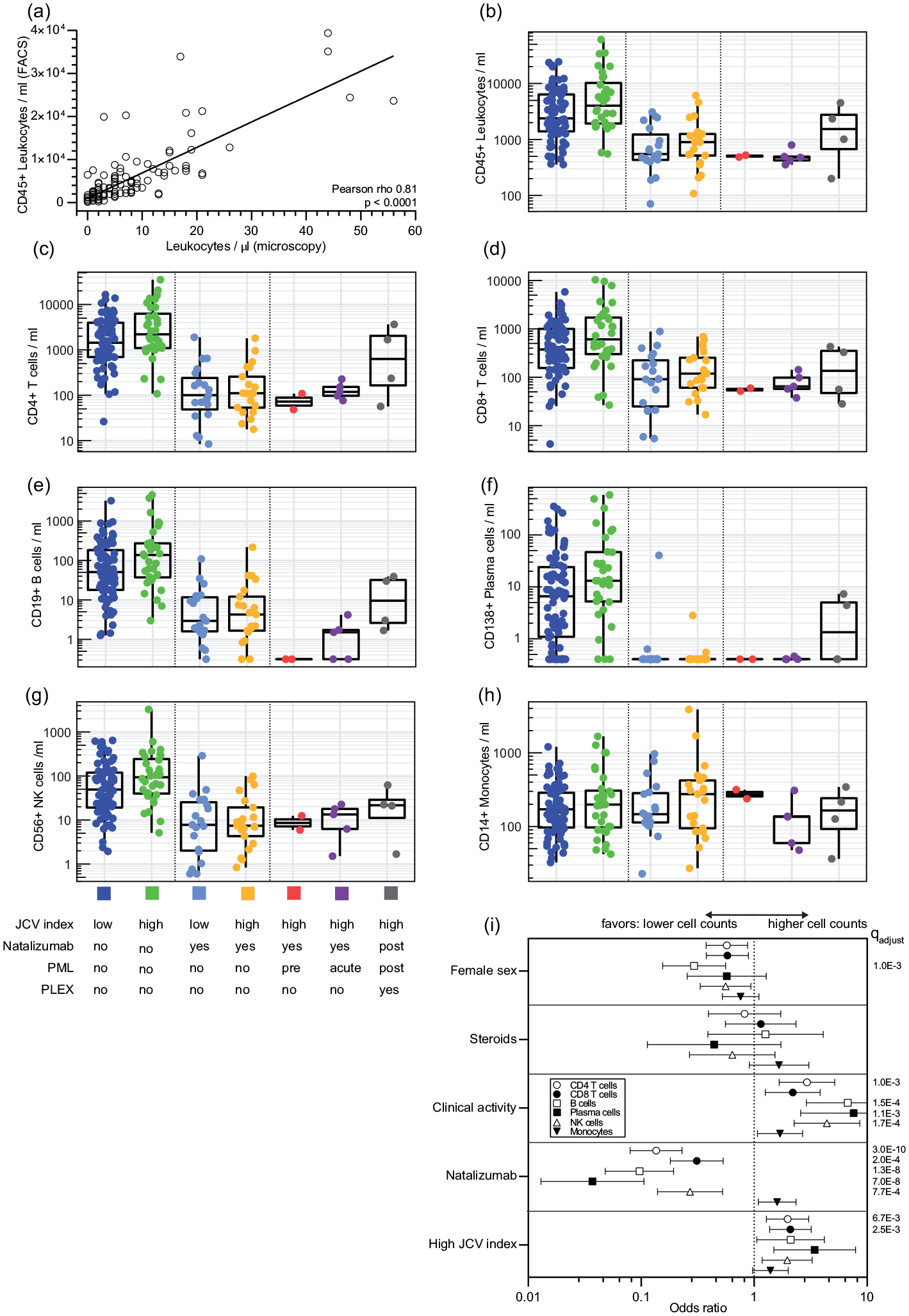
CSF immune cell subset counts: (a) CSF cell counts obtained by microscopy- or flow cytometry–based quantification are comparable. Shown are the values of 151 CSF samples with regard to leukocytes per µL (assessed using optical counting with a Fuchs–Rosenthal chamber) and CD45+ leukocytes per mL (assessed via flow cytometry (FACS) using reference beads). Both values correlate well (Pearson rho 0.8131), but there were 29 samples with a microscopy value of 0 cells/µL, while there were no values of 0 cells/mL in the flow cytometry assessment. The 29 samples with the microscopy value of 0 were measured with a median of 542.9 leukocytes/mL via flow cytometry. (b)–(h) Immune cell subset counts per mL of CSF as measured by FACS, according to ‘High JCV index’ and natalizumab treatment. Shown are boxplots based on unadjusted, raw data values with the middle line indicating the median and the box indicating the interquartile range (IQR; 25th and 75th quartile). Whiskers depict the data range excluding outliers with outliers defined as data points > 75th quartile + 1.5 × IQR or <25th quartile – 1.5 × IQR. PLEX = plasma exchange. (i) Forest plot illustrating the influence of the explanatory variables sex, steroid treatment, clinical activity, natalizumab treatment and ‘JCV index’ on the immune cell subset counts. The dotted line indicates the reference value for the binary variable (e.g. natalizumab versus non-natalizumab). Odds ratios (ORs) and p values were determined in multivariable generalized linear models using immune cell subset counts as the dependent variables and sex, age, disease duration, EDSS, clinical activity, steroid treatment, natalizumab treatment and JCV index as the explanatory variables. Type I error inflation was controlled by correcting the covariate-adjusted p values at 1% false discovery rate (FDR). FDR-controlled p values are declared as qadjust.
Results
While neither age, disease duration, EDSS, nor steroid treatment influenced CSF leukocyte counts, female sex was specifically associated with decreased B-cell counts. Natalizumab therapy strongly reduced CSF immune cell numbers, ranging from 69% (CD8 T cells, OR 0.31) to 96% (plasma cells, OR 0.04), while monocytes appeared unaffected. Conversely, clinical activity coincided with increased cell counts of B and plasma cells, and to a lesser extent CD4 and NK cells, while again monocyte CSF counts remained unchanged. To assess CNS immune cell abundance in the context of PML risk, anti-JCPyV serology was employed to divide patients into two PML risk categories: sero-negative or sero-positive patients with low anti-JCPyV antibody titers ( ‘JCV index’ < 1.5) in the ‘low risk’ category, and sero-positive patients with high anti-JCPyV antibody titers ( ‘JCV index’ > 1.5)2 in the ‘high-risk’ category. ‘JCV index’ above 1.5 was specifically associated with increased CD4 and CD8T-cell CSF counts, consistently in both untreated and natalizumab-treated patients.
Five patients with natalizumab-associated PML and high ‘JCV index’ values presented with comparably low CSF cell numbers, especially for CD8 and B-cells at the time point of PML diagnosis (acute). This seemed to precede the acute phase of viral encephalitis (pre), and appeared restored in three of four observations directly after plasma exchange (Figure 1(b)–(i)). However, due to the low number of available PML patient data, significance was not assessed for this group.
Discussion
We have indicated previously that monocyte abundance in the CSF is not affected by treatment with natalizumab. 15 Similarly, neither clinical activity, nor JCPyV serology was associated with alterations in monocyte counts, indicating that CSF-resident monocytes might not be continuously repopulated from the periphery. Plasma cells, which are known to be specifically present in CSF of relapsing MS patients were undetectable in 92% (35/38 non-PML patients +5/5 PML patients) of the assessed natalizumab-treated RRMS patients, indicating their involvement in MS disease pathology. Consistent with the treatment-associated reversal of the CSF CD4/CD8 ratio, 16 CD4 T cells seemed affected more strongly by natalizumab treatment than CD8 T cells indicating that remaining CD8+ T cells in CSF of natalizumab-treated patients originate from the CNS parenchyma-resident lymphocyte pool,11,17,18 while CD4 T cells are recruited to and surveil the CNS and CSF. 19 Accordingly, clinical activity was only associated with increasing CD4, but not CD8 T-cell numbers. The finding that patients with high PML risk show increased CSF cell counts indicates increased compartmentalized immune recruitment in these patients, presumably due to high viral activity. 10 It is also consistent with prior studies indicating that after an episode of viral activity CD4 T cells are sent to the CNS to help establish or support/maintain the tissue-resident immune defence orchestrated by CD8 T cells.11,12,20 –22 Similarly, in the absence of natalizumab, recruited plasma cells would contribute to intrathecal antibody production (and thereby MS pathology), and localized immune defence (supporting JCPyV defence).23,24 Predominantly CD8 tissue-resident memory cells (TRM) have recently been shown to reside in mucosal barriers but also tissues such as the CNS,17,18 to provide prolonged protection after the primary infection has been cleared.20,25,26 However, recent studies suggest that tissue-resident (polyoma) virus control by CD8 TRM needs constant help from infiltrating CD4 T cells.12,22 A failure, or prolonged reduction of immune recruitment to the site of viral activity (e.g. the gut or the CNS) might predispose for PML development,7,13 as evident from PML associated with either HIV infection or prolonged lymphopenia, and indicated by abnormal immune cell recruitment in the five included PML patients. While in patients with low ‘JCV index’ values (and low PML risk), localized JCPyV control might be sufficient, high anti-JCPyV antibody titers might indicate that patients would depend on repeated peripheral immune cell recruitment to support the local immune defence and control JCPyV.
This study suggests that high CSF cell counts, as observed in MS patients with clinical activity, could expectedly contribute to MS pathology, but also be part of systemic immune cell recruitment and compartmentalized surveillance against JCPyV. In consequence, lack thereof could be associated with increased PML risk. While the large overlap between PML and non-PML patients does not support CSF cell counts to be a clinically applicable risk biomarker, these findings support the hypothesis of excessively reduced immune cell recruitment to sites of viral activity, potentially also the CNS, as a contributing factor for the development of PML.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Tilman Schneider-Hohendorf received research and travel support from Novartis Pharma and Biogen. Andreas Schulte-Mecklenbeck received research and travel support from Novartis Pharma. Patrick Ostkamp and Claudia Janoschka received travel support from Novartis Pharma. Marc Pawlitzki received travel support from Novartis Pharma. Felix Luessi served on the advisory board of Roche and received travel funding from Teva. Frauke Zipp received funds for scientific consultation or research by DFG, BMBF, PMSA, Novartis, Octapharma, Merck Serono, ONO Pharma, Biogen, Genzyme, Celgene, Roche, and Sanofi-Aventis. Sven G. Meuth received honoraria for lecturing and travel expenses for attending meetings from Almirall, Amicus Therapeutics Germany, Bayer Healthcare, Biogen, Celgene, DiaMed, Genzyme, MedDay Pharmaceuticals, Merck Serono, Novartis, Novo Nordisk, Ono Pharma, Roche, Sanofi-Aventis, Chugai Pharma, QuintilesIMS, and Teva and received research support from Almirall, Amicus Therapeutics Germany, Biogen, DiaMed, Fresenius Medical Care, Genzyme, Merck Serono, Novartis, Ono Pharma, Roche, and Teva. Luisa Klotz received compensation for serving on Scientific Advisory Boards for Genzyme, Janssen, Merck, Novartis and Roche, received speaker honoraria and travel support from Novartis, Merck Sorono and CSL Behring and receives research support from Novartis and Biogen. Heinz Wiendl received honoraria and consultation fees from Bayer Healthcare, Biogen, Fresenius Medical Care, GlaxoSmithKline, GW Pharmaceuticals, Merck Serono, Novartis, Sanofi-Genzyme and TEVA Pharma. Catharina C. Gross received speaker honoraria from Mylan, Bayer Healthcare and Sanofi-Genzyme and travel expenses for attending meetings from Bayer Healthcare, Biogen, EUROIMMUN, Novartis and Sanofi-Genzyme. She received research support from Novartis and Biogen. Nicholas Schwab received travel and research support from Novartis and Biogen.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Deutsche Forschungsgesellschaft (DFG) Grant (grant no. CRC128) project B1 to NS, A09/A10/Z02 to HW, A09 to CCG, A08/Z02 to LK, the Kompetenznetz Multiple Sklerose (Competence Network for Multiple Sclerosis) funded by the Federal Ministry of Education and Research (FKZ 01GI1308B 01GI0907, and GER-TYS-12-10-401 (REGIMS)) to HW, and supported by Novartis Pharma GmbH to TSH. The funding agencies had no influence on design and conduct of the study; collection, management, analysis and interpretation of the data; and preparation, review or approval of the manuscript.